Ab-Initio Molecular Dynamics Simulations of Ultrafast Excited Graphite and Amorphous Carbon
 Bernd_Bauerhenne_Ab-initio molecular dynamics simulations of ultrafast excited graphite and amorphous carbon_Figure1
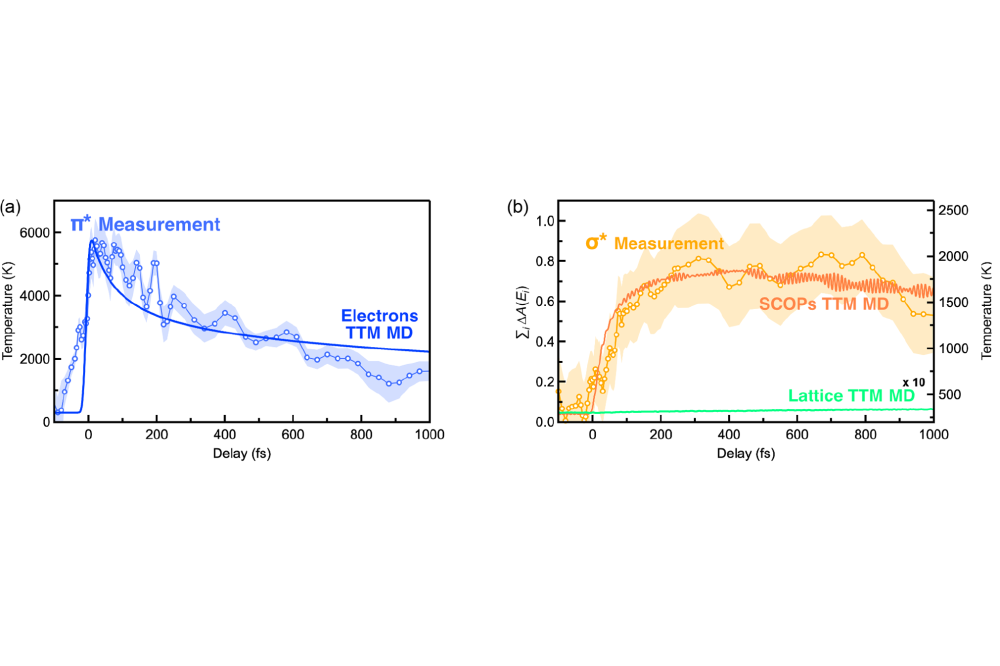
Bernd_Bauerhenne_Ab-initio molecular dynamics simulations of ultrafast excited graphite and amorphous carbon_Figure1Figure 1: The results from the MD simulation with the interatomic potential are compared with the experimental results retrieved from attosecond XANES for a femtosecond laser excitation of graphite. (a) The behavior of the electronic temperature is shown. (b) The behavior of the temperature of the SCOPs and the other phonon modes are shown. Figure is taken from Ref. [1], which is published under open access license.
Bernd Bauerhenne
Introduction
Femtosecond laser pulses excite the electrons of matter to a high temperature whereas the ions remain mainly unaffected. The generated state with hot electrons and cold ions is far away from thermodynamic equilibrium and exists for several picoseconds. Strong forces act on the ions and many ultrafast phenomena occur. Due to the electron-phonon coupling mechanism, the hot electrons transfer their energy to the lattice and cool down. Within this project, we performed molecular dynamics simulations of femtosecond laser pulse excited graphite, graphene and liquid carbon and compared directly with experiments.
Methods
We performed molecular dynamics (MD) simulations using temperature dependent density functional theory using small simulation cells. We created an electronic temperature dependent interatomic potential from part of these simulations. We performed large scale MD simulations using the new interatomic potential and analyzed the atomic coordinates.
Results
We performed MD simulations of bulk amorphous carbon, thin graphite films and graphene. The supercell for amorphous carbon contains 512 atoms with an atomic density of 2 g/cm2. The cell volume was kept constant. In order to initialize the structure, the atomic coordinates were set to a simple cubic structure and a MD simulation at an ionic temperature of 5000 K was performed using an Andersen thermostat. Within this simulation, the structure melts. Then, the structure was relaxed to obtain finally amorphous carbon. We simulated an excitation by a Gaussian shaped laser pulse of a full width at half maximum (FWHM) of 25 fs that leads to a final absorbed energy density of ~ 2.5 eV per atom. For this, we used electronic temperature dependent density functional theory and the two-temperature model. We used the value 17*1018 Wm-3K-1 for the electron-phonon coupling constant G in order to get the same equilibrium time of 0.3 ps like in the experiment, since there is no literature value of G for amorphous carbon. At first, the electronic temperature increases dramatically up to a maximal value of ~ 37000 K due to the laser excitation. Then the electronic temperature decreases and the ionic temperature increases due to the electron-phonon coupling until both temperatures reach the same equilibrium value of ~ 14000 K. We also analyzed the p electronic states during this processes and found out that they undergo a drastic rearrangement in less than 0.1 ps, consisting in a rise of the density of states across the Fermi level and a decrease 10-15 eV above that level. This agrees very well with the time-resolved x-ray absorption spectroscopy (TRXAS) measurements. In addition, we performed MD simulations of a thin graphite film using a supercell containing 252 atoms and of graphene using a supercell containing 180 atoms. We simulated several increased constant electronic temperatures. We used the obtained atomic forces and cohesive free energies from these MD simulations as reference data for fitting an electronic temperature dependent interatomic potential for laser-excited graphite. This interatomic potential describes accurately the phonon bandstructure, electronic specific heat, interatomic forces and energies for graphene and graphite. Then we performed MD simulations with the interatomic potential of a large graphite supercell containing 16128 atoms. We initialized the atomic coordinates and velocities at room temperature by applying an Andersen thermostat. A laser excitation by a 15 fs FWHM Gaussian shaped pulse was simulated by increasing accordingly the electronic temperature. Graphite exhibits a strong electron-phonon coupling between the electrons and a part of the phonons, the so called strongly coupled optical phonons (SCOPs). We modeled this effect by an individual electron-phonon coupling constant for the SCOPs. The electron-phonon coupling to the other phonon modes is very weak and was therefore neglected. We identified 980 phonon modes as SCOPs in our supercell. Due to the electron-phononn coupling, the electron temperature decreases and the temperature of the SCOPs increases significantly whereas the temperature of the other phonon modes remains almost unaffected. These findings agree very well with the experimental results obtained by attosecond core-level x-ray absorption finestructure spectroscopy (XANES), as it can be seen in Fig 1.
Discussion
We found an excellent agreement with the experimental results which validates our approach to simulate the effects of a femtosecond laser excitation on matter. The ability of creating highly accurate electronic temperature dependent interatomic potentials allows us to perform large scale MD simulations of laser excited matter. We will derive such potentials for further materials.