Computational Analysis of GPCR Biased Signaling
 Monteleone,Stefania-Computational_Analysis_of_GPCR_Biased_Signaling
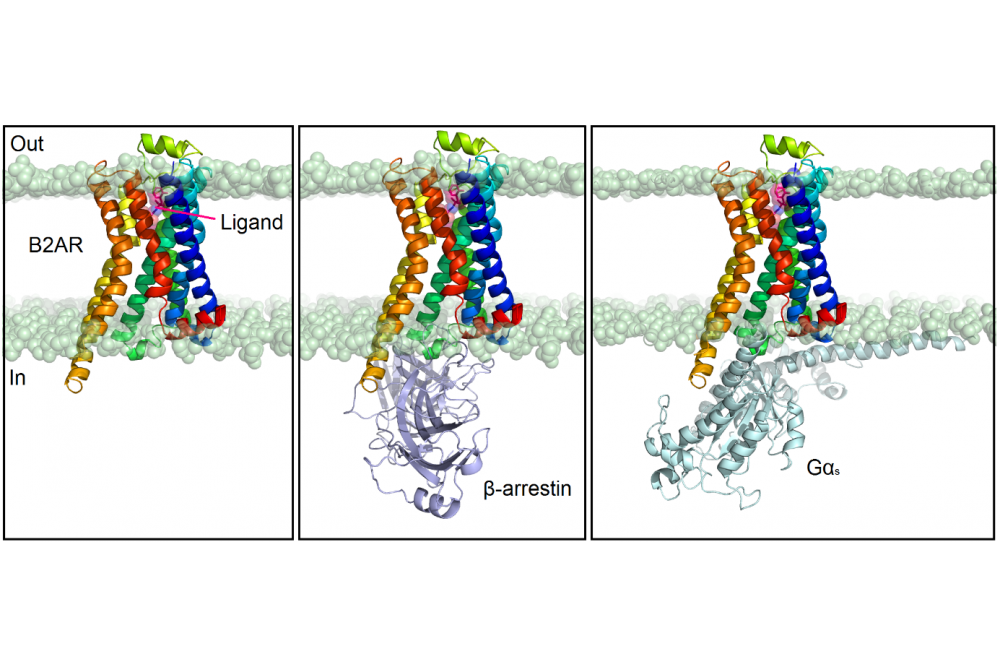
Monteleone,Stefania-Computational_Analysis_of_GPCR_Biased_SignalingFigure 1: Three distinct scenarios of systems that will be submitted to MD simulations for every ligand. The receptor (B2AR) is colored in rainbow and represented as cartoon. The ligand binds to the extracellular side, whereas the effectors (β-arrestin and G-protein) bind to the intracellular side of the receptor. The membrane is represented as spheres (only polar heads are displayed).
Stefania MonteleoneIntroduction
G protein-coupled receptors (GPCRs) constitute a large family of membrane proteins which share a conserved structural fold with seven transmembrane (TM) α-helices. They are involved in almost all cellular functions. About 30% of the drugs on the market target at least one GPCR, showing their key involvement in distinct human diseases [1]. For this reason, the structural understanding of their function plays an essential role in the design of new selective and safe drug candidates. Small molecules generally bind to the extracellular side of the receptor and generate a comparatively small structural rearrangement of the helical bundle in the ligand-binding pocket. This change is transmitted to the intracellular side of the receptor, which in contrast undergoes large conformational rearrangements that allow the receptor to bind cytoplasmic effector proteins such as G proteins and β-arrestins (Figure 1). Depending on the ligand, the receptor sometimes alters its preference for a particular cellular effector protein by predominantly interacting either with a G protein or a β-arrestin (β-arr): this mechanism is termed biased signaling [2]. A ligand that is biased may activate one G protein-driven pathway and recruit less β-arr, or vice versa. Consequently, biased GPCR ligands potentially are able to selectively target a specific cellular mechanism and might be capable of avoiding toxic side effects [3]. Yet, it is still not clear how precisely different ligand-receptor interactions translate to different conformational and activation states of a GPCR. Our goal is to understand, at the atomic level, the mechanism for Gs and β-arr recruitment bias at the β2-adrenergic receptor (B2AR), with the final purpose to design potent biased ligands.
Methods
All biomolecules are continuously moving under in vivo conditions. Protein structure dynamics represents the most crucial aspect in relation to molecular function, especially for GPCRs, which undergo conformational changes upon receptor activation. We apply homology modelling, molecular docking and molecular dynamics simulations to examine these structural changes of several ligand:receptor:effector complexes. This enables us to identify amino acids that are potentially responsible for biased signaling at the B2AR.
In particular, on the HPC cluster, we perform molecular dynamics (MD) simulations. This method describes the time evolution of a biomolecular system by solving the second Newton’s equation of motion. By computing the energy values with a defined force field, forces are derived with respect to the atomic positions. Forces are related to the acceleration and this allows computing the atomic velocities and new atomic positions of a biomolecule. Our systems are equilibrated with NAMD on Marc2 at Philipps-University of Marburg, which allows the use of multiple CPU cores. Afterwards, MD sampling is carried out on the Lichtenberg HPC cluster at the University of Darmstadt, with the program ACEMD, which takes advantage of GPU cards and uses a time step of 4fs, speeding up the calculations considerably.
Results
The analysis of several G protein-biased ligands which have recently been reported in literature [4] and the crystal structure of the B2AR in complex with a G protein of subtype Gs [5] allowed us to investigate the protein-ligand interactions and the structural conformation of the receptor over time upon binding of Gs-biased ligands.
In contrast, there is no experimental evidence for β-arr-biased ligands so far and no structures have been determined in complex with the B2AR. In this case, the design of potential biased ligands is more challenging and we analyzed the interactions between the B2AR and several unbiased ligands by performing MD simulations.
Results showed that the effector binding site changes conformation in order to contact the intracellular protein. Several interaction points have been identified via this simulation, but did not show significant differences between the Gs- and β-arr-bound simulations. However, we were able to identify different protein-ligand interactions in the orthosteric pocket. Here, small molecules bind to the receptor by forming strong interactions with conserved key amino acids. In some cases, additional hydrogen bonds or hydrophobic interactions might be responsible for triggering a specific conformation of the receptor such that it preferentially approaches Gs or β-arr.
Outlook
MD simulations represent a valid approach to understand the mechanism of GPCRs and the use of GPU cards on HPC clusters is fundamental to speed up the calculations. We plan to perform virtual screening of large libraries and simulate the top-ranked molecules to identify potential biased ligands based on the interactions with the protein.