Migration Barriers of Cations in Doped Lead-Free (Ba,Ca) (Zr,Ti) O2 Piezoceramics
 Paulik,Anna_Migration Barriers of Cations in Doped Lead-Free (Ba,Ca) (Zr,Ti) O2 Piezoceramics
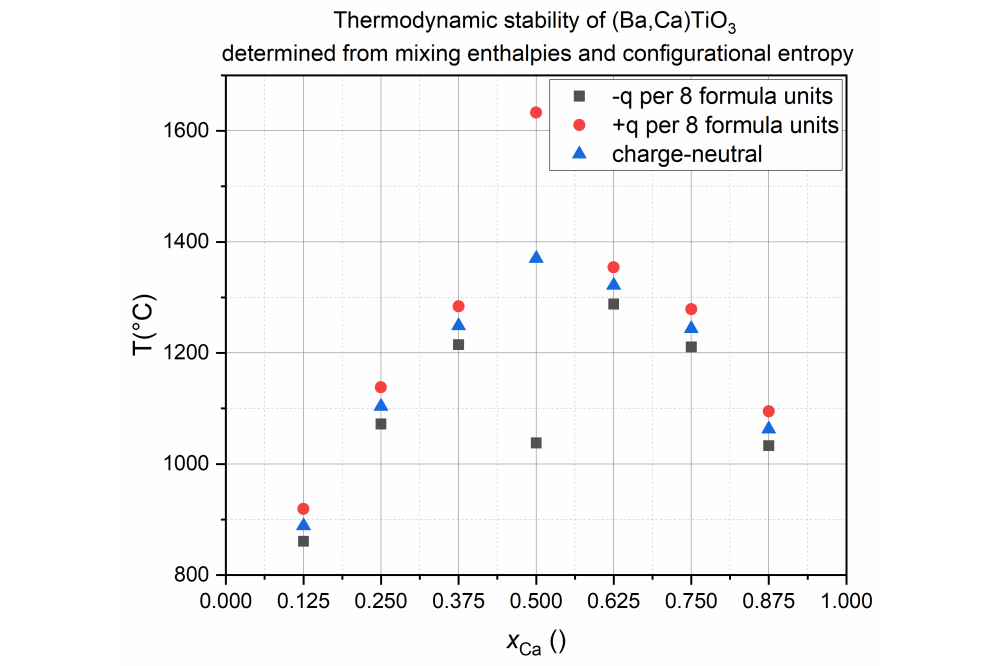
Paulik,Anna_Migration Barriers of Cations in Doped Lead-Free (Ba,Ca) (Zr,Ti) O2 PiezoceramicsThermal stability of a Barium Titanate - Calcium Titanate mixture, calculated from mixing enthalpies (DFT) and configurational entropy; depicted for a neutral structure, in comparison to a positively charged and negatively charged structure (realized by including one additional hole or electron per 2x2x2 supercell). Qualitative first results suggest that electronically compensated dopants may be used to increase or decrease miscibility at a given temperature.
Introduction
(Ba,Ca) (Zr,Ti) O3 (BCZT) is a lead-free piezoelectric material system that has been shown to exhibit outstanding electromechanical properties near its polymorphic phase boundary,making it suitable for applications in precision actuators, sensors and haptic devices. One of the main drawbacks of this solid solution is its large number of constituents, some of which exhibit slow interdiffusion. This leads to poor homogenization during synthesis as well as segregation of both matrix atoms and dopants, which are commonly added to improve the performance of the material. Furthermore, the thermodynamic stability of the BCZT solid solution is limited, which may also be influenced by the addition of dopants.
Methods
Density functional theory (DFT) calculations using the VASP (Vienna Ab initio Simulation Package) code were performed for different BCZT sub-systems (e.g., BaTiO3-CaTiO3). A 2x2x2 supercell was generated, and all non-energetically equal configurations of A-site cations were identified. The configurational enthalpy of the mixed systems was calculated, assuming either a linearly interpolated cell-size for the solid solutions based on Vegard’s law or the cell parameters of the individual perovskites for all solid solutions. Considering the calculated mixing enthalpies and entropic contributions, predictions for the miscibility of the systems at finite temperatures were made. The electronic band structures and density of states were calculated. Furthermore, acceptor and donor-doped subsystems were simulated by changing the total electronic charge of the supercell (mimicking the electronic compensation of an acceptor- or donor-dopant atom). The changes in the thermodynamic stability and band structure were investigated.
Results
We found that both the (Ba,Ca)TiO3 and Ba(Zr,Ti)O3 subsystems have a positive enthalpy of mixing if linearly interpolated lattice constants are assumed, suggesting that no solid solution is formed. This is experimentally confirmed for the (Ba,Ca)TiO3 subsystem. We compared the calculated Gibbs’ free energy to the BaTiO3-CaTiO phase diagram, published in 1955 by DeVries and Roy, and found good qualitative agreement. Notably, the enthalpy of mixing was negative for all solid solutions of the Ba(Zr,Ti)O3 subsystem when assuming fixed lattice parameters (of either BaTiO3 or BaZrO3) instead of interpolation. Experimentally, no miscibility gap is observed in the Ba(Zr,Ti)O3 system, suggesting that mixing on one host lattice without significant changes of the lattice parameters might be more favorable. However, it has to be noted that kinetic effects may also influence the apparent miscibility of the systems.
Discussion
The simulated doping through the addition or subtraction of electronic charges (essentially artificially shifting the Fermi level) systematically influenced the mixing enthalpy of the systems. Donor doping slightly increased the miscibility while acceptor doping had the opposite effect (with an outlier of Ba0.5Ca0.5TiO3, where this effect was significantly exacerbated). This trend could not be directly replicated experimentally, since acceptor- or donor-dopants have different ionic radii than the lattice atoms, leading to lattice strains, and are frequently ionically compensated. Nevertheless, the performed simulations give important theoretical insights into the expected thermodynamic stability of the complex BCZT solid-solution. To increase the practical relevance of the simulation results for doped systems, one would need to implement Fe3+ or Nb5+ atoms on the Ti4+ or Zr4+ sites, as well as compensating oxygen vacancies. This significantly adds to the complexity of the system and increases the computational costs. Nevertheless, further experimental insights regarding the types of expected defects may help to define less computationally demanding questions, which may be answered with similar thermodynamic calculations