Computational Stability Investigations on High Entropy Oxides for Oxygen Evolution Electro-Catalysis
 Vipin,Kumar_Computational Stability Investigations on High Entropy Oxides for Oxygen Evolution Electro-Catalysis_Figure1
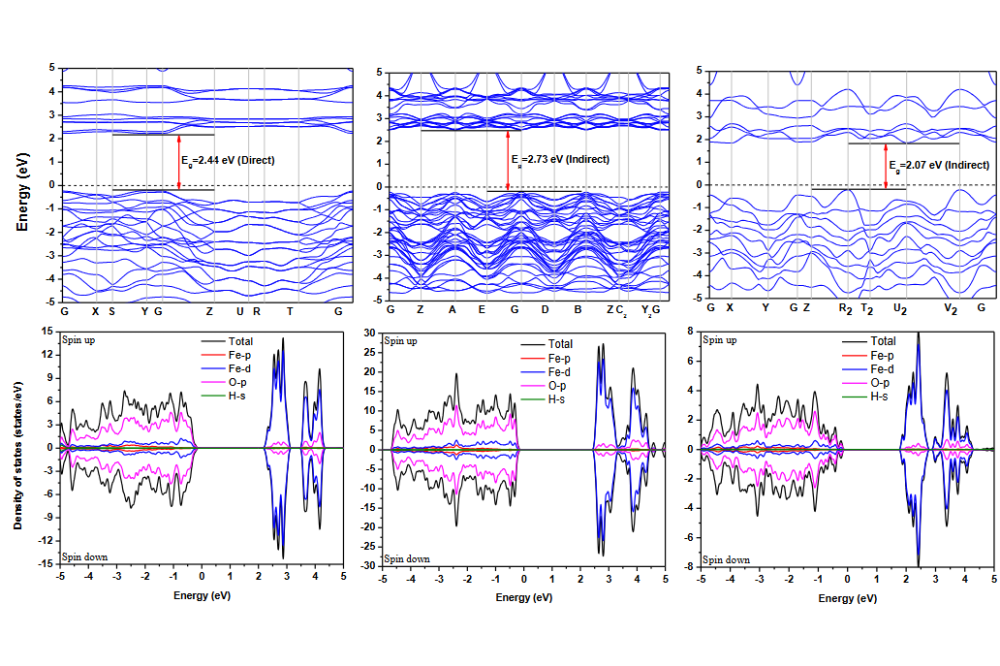
Vipin,Kumar_Computational Stability Investigations on High Entropy Oxides for Oxygen Evolution Electro-Catalysis_Figure1Figure 1: The electronic band structure and partial density states (PDOS) of iron oxyhydrides FeOOH (a) goethite, (b) akageneite, and (c) lepidocrocite.
 Vipin,Kumar_Computational Stability Investigations on High Entropy Oxides for Oxygen Evolution Electro-Catalysis_Figure2
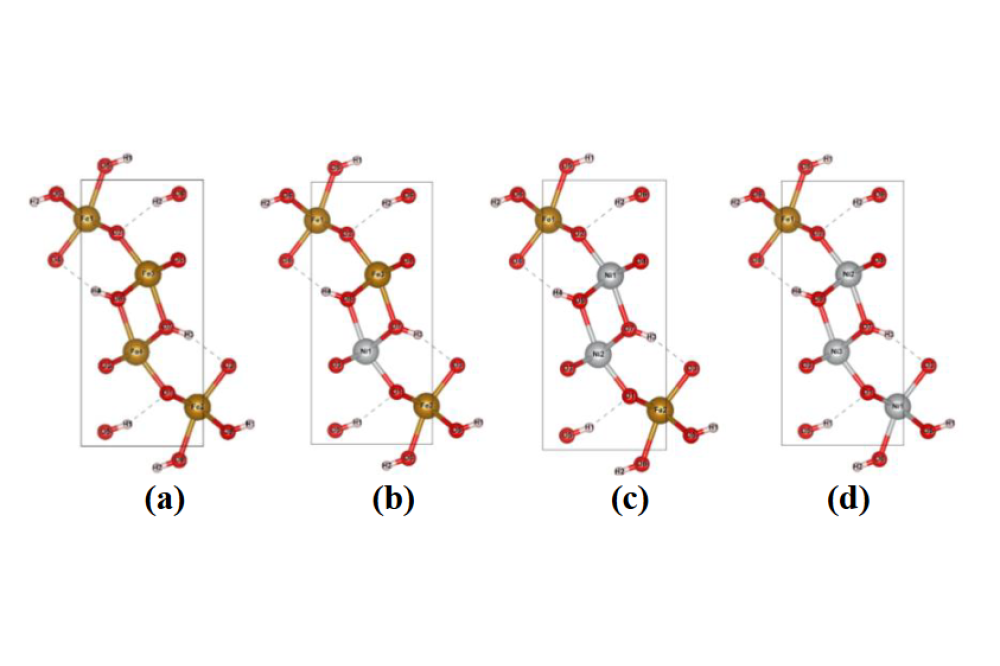
Vipin,Kumar_Computational Stability Investigations on High Entropy Oxides for Oxygen Evolution Electro-Catalysis_Figure2Figure 2: Crystal structure of substituted Ni2+ in (a) α-FeOOH, (b) Fe3Ni1OOH (c) Fe2Ni2OOH, and (d) Fe1Ni3OOH; where red, brown and grey color denote to O, Fe, Ni atoms.
 Vipin,Kumar_Computational Stability Investigations on High Entropy Oxides for Oxygen Evolution Electro-Catalysis_Figure3
Vipin,Kumar_Computational Stability Investigations on High Entropy Oxides for Oxygen Evolution Electro-Catalysis_Figure3Figure 3: Fe0.2Co0.2Mn0.2Cr0.2Ni0.2OOH structure generate by python script with randomly arrangements of Fe, Mn, Cr, Ni, Co element in α-FeOOH.
Introduction
High-entropy alloys (HEAs) materials are well-defined by their formation of at least five or more principal elements in nearly equimolar proportion. These unique newly compositional emerged as promising materials due to their fascinating physicochemical properties and multi-functionality. In contrast to conventional alloys, the intrinsic complexity of high-entropy alloys (HEAs) and oxyhydroxides arises from the substantial configurational entropy, which helps stabilize single-phase structures despite substantial atomic radii and electronegativity differences among constituent elements. Recent studies show that high-entropy oxyhydroxides (HEOHs), show outstanding catalytic activities due to the synergistic interactions among multiple metallic elements. These interactions modify the local electronic environment, optimize adsorption energies of catalytic intermediates (i.e. O*, OH*, OOH*, etc.), and encourage superior catalytic performance compared to traditional single-element or binary systems. For example, single-crystal nanoporous high-entropy (oxy)hydroxides have confirmed enhanced electrocatalytic activities attributed to structural and electronic synergistic effects arising from their high configurational entropy. Computational approaches using density functional theory (DFT), have significantly contributed to explaining the electronic and structural mechanisms governing these improvements. DFT studies on hydrogen storage in HEAs revealed that element-specific electronic interactions intensely affect bonding characteristics and thermodynamic stabilities of the materials. Similarly, computational modeling of lattice distortions in single-phase HEAs has offered important insights, revealing how atomic interactions and charge redistribution help reduce lattice strain and improve phase stability-factors which play a key role in enhancing catalytic performance. Additionally, computational investigation approach indicates that charge transfer and local lattice distortions within multi-component alloys play critical roles in stabilizing single-phase structures and optimizing their electro-chemical properties. The fine-tuned balance of charge distribution across multiple elements significantly reduces atomic size mismatch, resulting in reduced structural defects and improved catalytic activities. In the present project report, we utilized DFT calculations to explore the formation energetics, stability, and electronic structure of novel high-entropy oxyhydroxides. Our primary goal is to understand how varying elemental compositions effect the structural stability and electronic properties essential for electrocatalytic performance. Therefore, we aim to support the rational design of high-efficient, entropy-stabilized catalytic materials.
Methods
Density Functional Theory (DFT) investigation are performed using VASP (Vienna Ab Initio Simulation Package) code. The Projector augmented-wave (PAW) potentials were used as provided with VASP and the Perdew-Burke-Ernzerhof (PBE) gradient approximation for the exchange-correlation functional. The semi-core 3p/4p/5p electrons of Fe, Mn, Cr, Ni, and Co are explicitly treated as valence. The Brillouin zone sampling were performed, using the Monkhorst-Pack k-point meshes scheme. The crystal structure was visualized using the VESTA visualization code.
Results
We investigated the periodic crystal structures of three FeOOH polymorphs: (a) α-FeOOH (goethite), (b) β-FeOOH (akaganeite), and (c) γ-FeOOH (lepidocrocite). Fig. 1 shows the electronic band structure along high symmetry K-path and partial DOS for all three phases. The calculations show that α-FeOOH exhibits a direct band gap of 2.44 eV, whereas β-FeOOH and γ-FeOOH display indirect band gaps of 2.72 eV and 2.07 eV, respectively. Based on these findings, α-FeOOH was selected as the host structure for further high-entropy oxyhydroxide(HEOH) investigations. Fig. 2 presents Ni²⁺-substituted binary configurations at 1:1, 3:1, and 1:3 Fe/Ni ratios within the α-FeOOH unit cell (4 Fe, 8 O, and 4 H atoms). The results indicate that Ni preferentially adopts a +2 oxidation state rather than +3, which reduces the stable degree of protonation. Structural stabilities were estimated by referencing the energetics of the corresponding oxides. Table 1 summarizes the net charges in the system following Ni²⁺ substitution, confirming that protonation restores overall charge neutrality. The same substitution and charge-compensation procedure was applied to Fe³⁺, Mn³⁺, Cr³⁺, and Co²⁺ dopants. Afterward, a Python script was used to generate random high-entropy oxyhydroxide configurations. Fig. 3 shows representative examples of these randomly generated structures. The lowest-energy configuration among these will be selected for detailed thermodynamic analysis, which is currently underway.
| Compound | Ratio | Total Charges |
| FeOOH | 4:0 | 0 |
| Fe0.75Ni0.25OOH | 3:1 | -1 |
| Fe0.50Ni0.50OOH | 2:2 | -2 |
| Fe0.25Ni0.75OOH | 1:3 | -3 |
Discussion
The electronic-structure analysis of FeOOH polymorphs highlights α-FeOOH as the most suitable host for high-entropy design. Its observed to be a direct band gap, higher thermodynamic stability, and well-defined octahedral framework which offer a robust structural platform for multi-cation substitution. In contrast, β-FeOOH and γ-FeOOH refer to lower structural stability and partial cation occupancy, making them less favorable for high-entropy applications. Ni²⁺ substitution in α-FeOOH reveals charge-compensation nature consistent with the oxyhydroxide chemistry: protonation adjusts to maintain electroneutrality upon incorporating mixed-valence cations. This mechanism is critical for allowing a broad compositional idea in HEOHs. The consistent application of the same methodology to Fe³⁺, Mn³⁺, Cr³⁺, and Co²⁺ confirms that α-FeOOH can accommodate a choice of transition-metal cations with minimal structural disruption. The generation of random high-entropy configurations via the Python script workflow offers a statistically various structural ensemble, allowing identification of the lowest-energy structure. Fig. 3 is random high-entropy configurations which was generated by Python script workflow. This approach supports systematic exploration of configurational entropy effects and phase stability in emerging high-entropy oxyhydroxides. The ongoing thermodynamic screening will establish structure-property correlations necessary for future catalytic or functional applications.