Spectroscopic Properties and ORR Reaction Paths for FeNC Catalyst Models
 Gallenkamp_Charlotte_Spectroscopic Properties and ORR Reaction Paths for FeNC Catalyst Models_Fig1
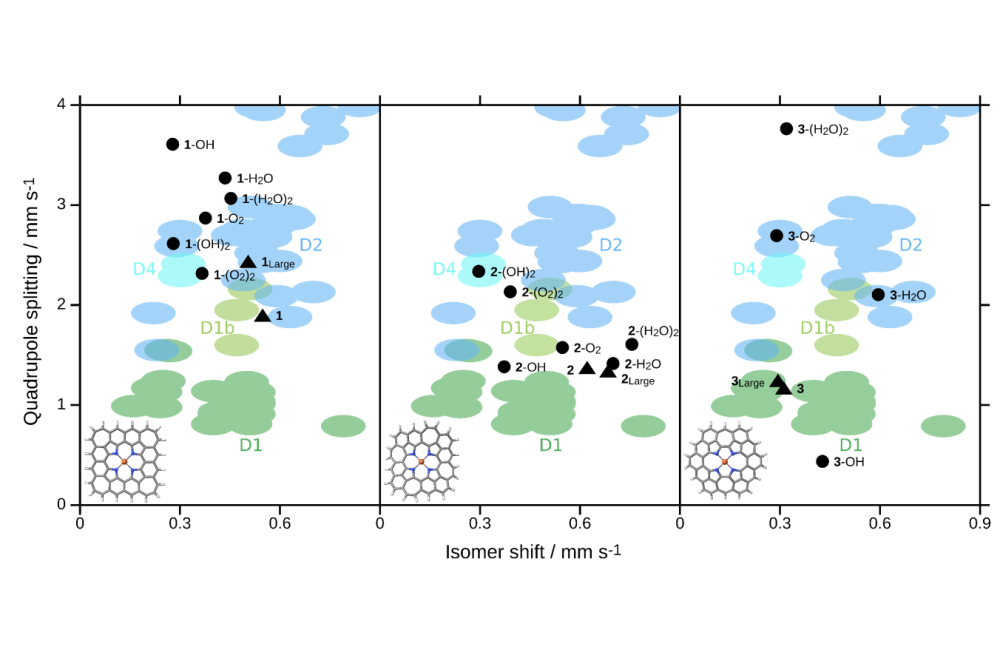
Gallenkamp_Charlotte_Spectroscopic Properties and ORR Reaction Paths for FeNC Catalyst Models_Fig1Figure 1: Mössbauer parameters, namely isomer shift and quadrupole splitting, of several tetrapyrrolic FeNC model complexes calculated using density functional theory (black) together with experimental Mössbauer parameters taken from various literature sources (colored ellipsoids). The model investigated is shown in the lower left corner of the corresponding plot. Triangles indicate models without further ligands, whereby the “large” model has an expanded graphene plane. Circles indicate models with intermediates of the oxygen reduction reaction as axial ligands, as denoted by the labels. The color of the ellipsoids represents the allocation to typical experimental FeNC signals, whereas their size illustrates the error in the computed Mössbauer parameters as determined by an in-house calibration study.
Niklas von RheinIntroduction
With street traffic being a major contributor to carbon dioxide emission and global warming, alternatives to the combustion engine currently in use are needed. Aside from electric vehicles, hydrogen powered fuel cells are a promising technology. Their high platinum load, however, stands in the way of general applicability. So-called FeNC catalysts, iron and nitrogen doped carbon materials, are promising for replacing platinum catalysts for one of the reactions in the fuel cell, the oxygen reduction reaction. Therefore, they are a widely researched field.
FeNC catalysts are prepared using pyrolysis, making them amorphous materials. Accordingly, the exact structure of the catalytically active center is not known, hampering a systematic improvement of this catalyst type. The amorphous structure necessitates the use of nucleus specific spectroscopy methods for structural elucidation. The most common method is Mössbauer spectroscopy, which needs to be referenced against mostly theoretical model complexes. The closer these models are to the real structure of the catalytic center, the more information they can yield.
In this project, we develop and test new FeNC models, calculate their Mössbauer parameters using quantum chemistry and compare them to experimental Mössbauer data. Our models differ in the shape of the catalytic center and the size of the carbon plane surrounding it.
Methods
We apply density functional theory for quantum chemical calculations, one of the most common methods in quantum chemistry. In density functional theory, the electronic energy is calculated using the electron density, which is connected to the energy by a functional. The exact form of the functional is not known but can be approximated.
Results
Mössbauer parameters were calculated for several types of model complexes. By varying the size of the carbon plane for two catalytic centers with different charges, a sensible model size was determined, being a good compromise between accuracy and computational cost.
Furthermore, for three tetrapyrrolic models, Mössbauer parameters have been calculated with the intermediates appearing in the oxygen reduction reaction as axial ligands. These have been compared to experimental Mössbauer parameters from a broad range of publications. Also, the covalency of their metal ligand bonds was investigated. However, even though the covalency changed significantly between the models due to the different degrees of planarity, it was not possible to connect the covalency to the Mössbauer parameters in a qualitative way.
Discussion
We showed that two of the three tetrapyrrolic models – a planar and a concave non-planar one – could explain most of the common Mössbauer signals in FeNC catalysts. Despite the different coordination geometries, the calculated Mössbauer parameters are similar for structures with the same electronic ground state. However, the differences in planarity and bond distances can lead to stabilization of different molecular orbitals and thus different ground states and Mössbauer parameters.
As no quantitative correlation between the covalency and the Mössbauer parameters could be obtained, the importance of using a broad range of model complexes with different coordination spheres around the iron atoms is emphasized.