Wigner Sampling of Mössbauer-Parameters
 von_Rhein_Niklas_Wigner Sampling of Mössbauer-Parameters_Fig1
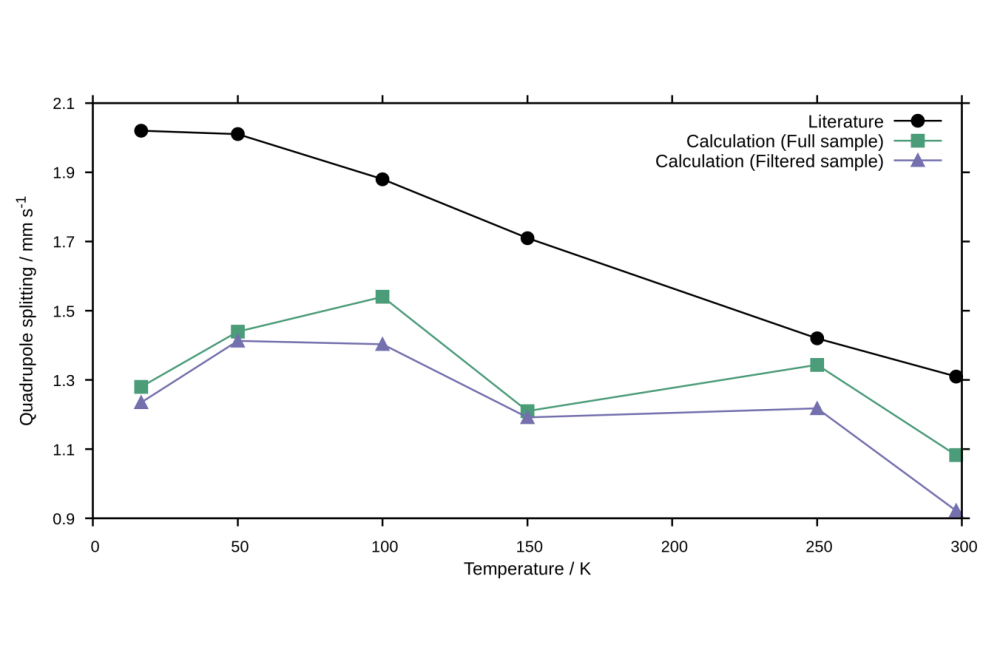
von_Rhein_Niklas_Wigner Sampling of Mössbauer-Parameters_Fig1Figure 1: Temperature behavior of the [Fe(OEP)(2-MeHIm)]-complex’ quadrupole splitting. In black, experimental literature data is shown. Green and purple indicate data obtained by our novel approach, using all structures in the created ensemble (green) or a filtered ensemble with only the dominant component (purple).
Niklas von RheinIntroduction
Mössbauer spectroscopy is a powerful tool for investigating iron in molecular and especially amorphous systems. Combined with quantum chemical calculations, it promises to enable structural elucidation for various relevant compounds such as FeNC catalysts. However, like many spectroscopy methods, it is temperature dependent, hampering the correlation of experiments at broad temperature ranges to quantum chemical calculations assuming zero Kelvin. Our group is developing a sampling-based method to access one contribution to the temperature dependence of Mössbauer parameters: The effect of molecular vibrations. This is not only promising for accessing the temperature dependence of one of the Mössbauer parameters, the quadrupole splitting, but can also help in gaining a deeper understanding of the relationship between quadrupole splitting and structural and electronic properties. In this project, the novel method was tested on a single structure (the [Fe(TPP)(2-MeHIm)]-complex seen in figure 1), for which experimental Mössbauer parameters at various temperatures are available in literature. This was done within the context of a student internship, combining scientific research with didactic teaching of the use of high-performance computing in theoretical and quantum chemistry.
The complex investigated, ([Fe(OEP)(2-MeHIm)]), exceeds 50 atoms and therefore large computational resources are needed for obtaining relevant results. Furthermore, calculations have to be performed for every structure in the created ensemble, while a large ensemble size of at least 600 structures is necessary for the method. Thus, a high-performance cluster is needed to carry out the calculations.
Methods
Quantum chemical calculations were performed using the density functional theory approach, a common method used in quantum chemistry connecting the electronic density to the energy of the system. Ensembles of vibrationally distorted structures were created with Wigner sampling, a sampling approach using the vibrational modes of the system investigated.
Results
At five temperatures, Mössbauer parameters have been calculated for ensembles of 600 structures and at one temperature for an ensemble of 900 structures. By averaging over all structures in one ensemble, temperature dependent Mössbauer parameters are calculated.
Discussion
For the given structure, the method struggles to reproduce not only the exact experimental results, but also the expected trend. A reason is seen in the occurrence of different electronic states due to deformation, making the approach highly dependent on the quantum chemical method used. This is supported by the observation that upon considering only data points of the dominant electronic state, the correlation between experiment and theory can be improved.
Viewing the Mössbauer parameters of all structures in the ensemble, it is observed that the sign of the quadrupole splitting can shift upon deformation with the absolute value remaining similar. This matches the expectations, as the quadrupole splitting is highly dependent on the shape of the charge distribution around the iron nucleus, and this shape changes upon vibrational deformation. Therefore, this approach gives a better understanding of the vibrational temperature effect, as it makes this sign swap upon deformation visible.
Using the large number of structures within the ensemble, we furthermore aimed to gain a better understanding of the Mössbauer parameters by correlating them with structural parameters. A particular focus was placed on the dependence of the Mössbauer parameters on the movement along certain normal modes. By a linear combination of the normal modes, nuclear movements correlating strongly to the Mössbauer parameters can be calculated. These are analogous to the movements in a structurally similar complex investigated beforehand and consistent with chemical expectations.
Outlook
Future works will include testing the novel approach on more systems, to identify and overcome errors and to benchmark it more precisely. Also, the use of different sampling techniques is promising as well as the use of quantum chemical methods beyond density functional theory to assess and possibly overcome the method dependence of the approach.