Testing a Sampling Based Method for Capturing the Mössbauer Spectroscopy’s Temperature Dependence
 von_Rhein_Niklas_Testing a Sampling Based Method for Capturing the Mössbauer Spectroscopy’s Temperature Dependence_Fig1
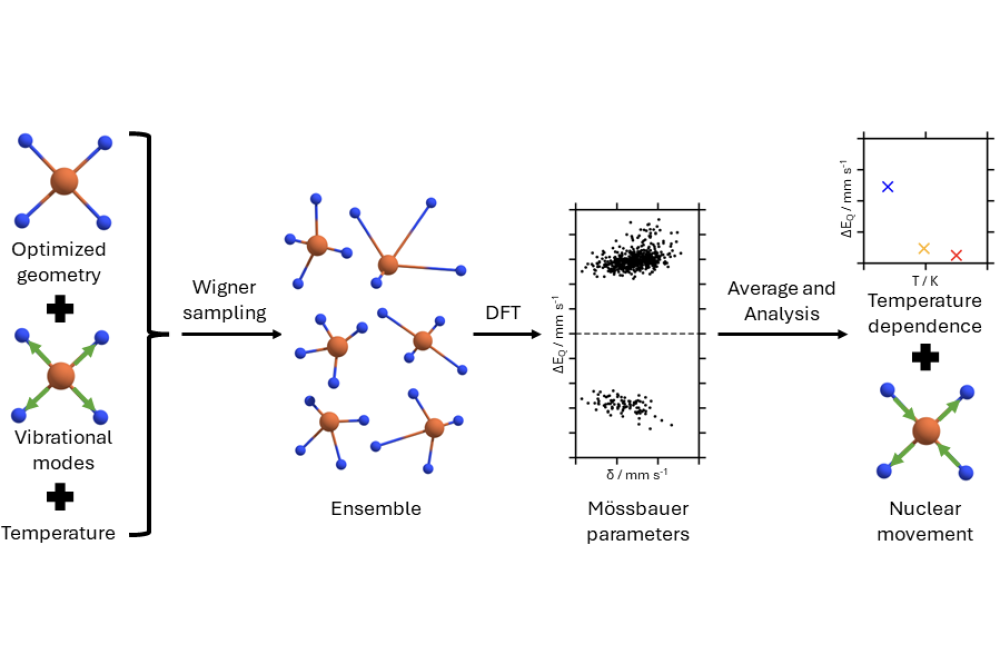
von_Rhein_Niklas_Testing a Sampling Based Method for Capturing the Mössbauer Spectroscopy’s Temperature Dependence_Fig1Figure 1: Schematic representation of the sampling procedure used to calculate temperature dependent Mössbauer parameters. First, with the optimized geometry and the vibrational modes of the investigated complex, Wigner sampling is used to create an ensemble of structures at a given temperature. Then, the Mössbauer parameters, namely isomer shift (δ) and quadrupole splitting (ΔEQ), are calculated for each structure using density functional theory (DFT). Finally, the temperature dependent Mössbauer parameters are determined by averaging the given parameter over all structures in the ensemble. Furthermore, using a normal mode analysis approach, a nuclear movement with high correlation to the Mössbauer parameters is computed.
Niklas von RheinIntroduction
Combined with quantum chemical calculations, Mössbauer spectroscopy is a powerful tool to elucidate the structure of molecular complexes and even iron compounds in amorphous materials like FeNC catalysts. However, comparison between experiment and theory is hampered by its temperature dependence: Quantum chemical calculations are formally performed at zero Kelvin, making experimentally demanding low temperature Mössbauer measurements necessary for a meaningful comparison of theory and experiment. We are developing a method to compute one aspect of temperature dependency of the Mössbauer parameters and among them especially the quadrupole splitting: vibrational distortion. Our sampling-based approach promises not only an estimation of the quadrupole splitting’s temperature dependence but even a better understanding of the Mössbauer parameters themselves, especially their correlation to electronic and structural parameters. Temperature dependent experimental Mössbauer parameters are mainly measured for complexes of either catalytic or biological relevance, most of which exceed 50 atoms. This is also true for the complexes used to test the newly developed method so far, making the quantum chemical calculations computationally demanding. Furthermore, large ensembles of structures (600 single-point calculations) are needed for meaningful results. Both aspects necessitate the use of a high-performance cluster.
Methods
Density functional theory, a widespread approach, was used for quantum chemical calculations. In density functional theory, the electron density is used to calculate the electronic energy of the system.
Wigner sampling was used for creating ensembles of vibrationally distorted structures representative for each temperature. It uses the vibrational modes of the initial structure for the distortion.
Results
Mössbauer parameters have been calculated for three complexes at three temperatures with ensemble sizes of 600 structures. Temperature dependent Mössbauer parameters were computed using the average over all structures in one ensemble.
Discussion
It was shown that our new approach is able to reproduce the temperature dependence of one Mössbauer parameter, the quadrupole splitting, qualitatively, though not quantitatively. One reason for the differences between prediction and experiment is seen in the possible reordering of molecular orbitals due to deformation and the high dependence on the
quantum chemical method this effect introduces.
Even though our approach is hampered by this method dependency, it gave valuable insights into the reason for the quadrupole splitting’s temperature behavior: The sign of the quadrupole splitting depends on the shape of the charge distribution and therefore a structural deformation can lead to a sign swap. The number of those sign swaps changes with the temperature due to the population of different vibrational states.
With a large number of vibrationally distorted structures at hand, we also investigated the correlation of the Mössbauer parameters to the displacement along the normal modes. By doing so, it was possible to calculate a nuclear movement with a large correlation to the Mössbauer parameters, giving rise to an even better understanding of the Mössbauer parameters’ dependence on the molecular structure.
Outlook
In future works, we will test and benchmark the approach on more systems and identify and overcome errors still prevailing. Furthermore, other methods for both quantum chemical calculations and sampling will be tested.