Towards a Mobility Preserving Coarse-Grained Model: A Data Driven Approach
 Saientan Bag_Towards a Mobility Preserving Coarse-Grained Model: A Data Driven Approach_Figure1
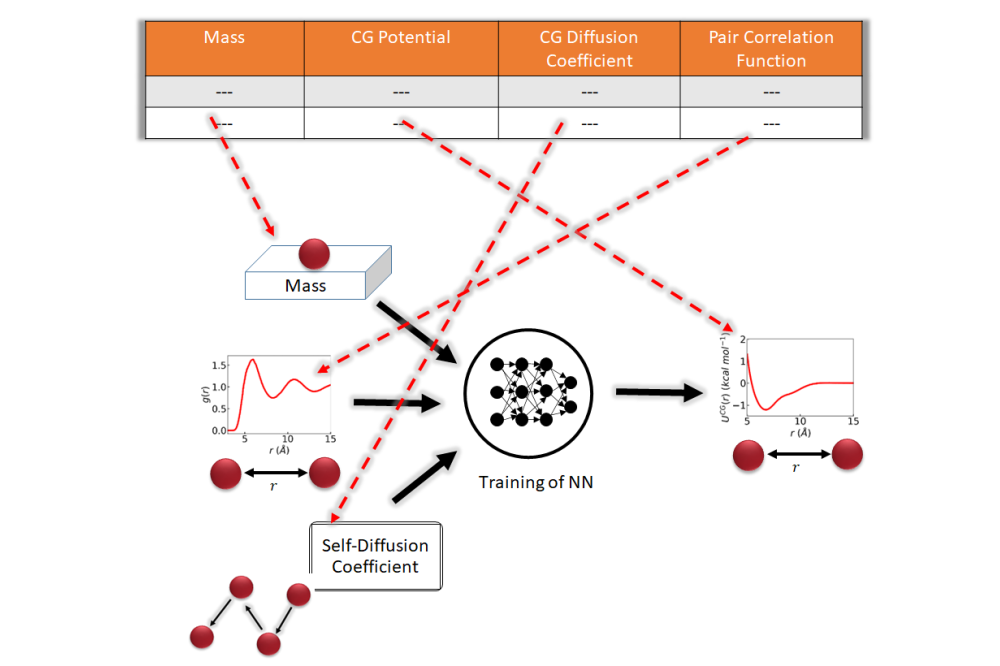
Saientan Bag_Towards a Mobility Preserving Coarse-Grained Model: A Data Driven Approach_Figure1Figure 1: A neural network is trained to predict mobility-preserving coarse-grained potential where the mass of the coarse-grained bead, the coarse-grained diffusion coefficient and the pair-correlation function are given as input.
Saientan BagIntroduction
Coarse-grained molecular dynamics (MD) simulation provides a faster alternative to all-atom MD simulation, essential for fast calculation of system properties. Therefore, in the last decade there has been continuous development of coarse-grained model. In spite of these attempts, the available coarse-grained models in the literature share a major drawback by introducing an artificial acceleration in molecular mobility. This makes the CG simulation unreliable in describing the dynamical processes in the systems both quantitatively and qualitatively. With the goal of solving this long-standing problem, in this manuscript we report a data driven approach to generate coarse-grained models, which preserve the all-atom molecular mobility. While the scientific community has warmly embraced the development of data-driven models, the pivotal role of a robust dataset to train these models often goes unnoticed. To deepen our understanding of physical systems and advance methodologies through data-driven approaches, the dataset itself should not be confined to physically realistic models. In this article, we leverage this insight by generating synthetic data to train a machine learning model. Subsequently, we employ this trained model to predict a mobility-preserving coarse-grained model for physical liquids. A huge amount of classical molecular dynamics simulations needs to be performed to generate the database and therefore the use of HPC is required.
Methods
To create a database of coarse-grained potentials, we exploit the fact that the all-atom reference model need not actually represent a molecule physically viable in nature. The synthetic database is built by changing the bonded all-atom force field parameters of three molecules: 2,3,4-Trimethylpentane (234TriMePe), Ethylbenzene (EtBz) and 3-Methylpentane (3MePe). We first performed all-atom molecular dynamics simulations for many different random choices of the bonded force-field parameters of the three molecules mentioned above. We further performed Iterative Boltzmann Inversion to generate coarse-grained potential corresponding to each all-atom molecular dynamics run. Coarse-Grained molecular dynamics simulations are performed in the end with the Iterative Boltzmann Inversion generated coarse-grained potential. For greater diversity of our database, we randomly choose one of the generated CG potentials and randomly assign it a bead mass between 20 to 200 (g/mol) and perform a CG simulation. We repeated (random selection of potential and random selection of mass) this procedure many times and performed new CG simulations. With the generated database we trained a neural network to predict mobility preserving coarse-grained models.
Results
With the database in hand, we designed the machine learning model (ML) which attempts to generate both structure and dynamics conserving CG potentials of a given all-atom model. To elaborate, we trained a NN to predict the CG potential given the mass of the CG bead, the pair correlation function and self-diffusion coefficient of the beads as input. We then use the trained NN to predict the CG potential of a pristine all-atom model given its mass, its center-of-mass pair correlation function and its atomistic self-diffusion coefficient. When we compare our machine learning-based coarse-grained potential with the one from iterative Boltzmann inversion, the machine learning prediction turns out better for all eight hydrocarbon liquids we studied. As all-atom surfaces turn more non-spherical, both ways of coarse-graining degrade. Still, the neural network outperforms iterative Boltzmann inversion in constructing good quality coarse-grained models for such cases. The synthetic database and the developed machine learning models are freely available to the community, and we believe that our approach will generate interest in efficiently deriving accurate coarse-grained models for liquids.
Discussion
All coarse-grained potentials exhibit limitations in accurately preserving the structure and dynamics of the all-atom model simultaneously. The ML predicted CG models strike a balance between structure and mobility. While this approach to generate potential is not perfect, it currently represents the best available option as, to the best of our knowledge, neither physics-based nor data-driven methods exist for predicting the structure and dynamic preserving potential. The reason might quite possibly be that such universal coarse-grained models do not exist. Both the database and the capabilities of the machine learning models exhibit certain limitations. Specifically, the database was constructed under fixed temperature and pressure conditions, while the coarse-grained model employed a single bead representation. It is important to emphasize that this work does not represent the final stage in the development of coarse-grained models, nor does it mark the end of progress in this field. On the contrary, the database itself is designed to be extendable and reusable. As an illustration, in our previous research, this database solely encompassed the 2,3,4-Trimethylpentane system. However, in this study, we have expanded its scope and generalized the machine learning models to predict the coarse-grained potential of all-atom models across considerably wider ranges. Furthermore, we have made this database freely available, encouraging its use and extension for the development of alternative models.