A Generic 3D-Diverse Fragment Library for Crystallographic Screening and Drug Discovery
Introduction
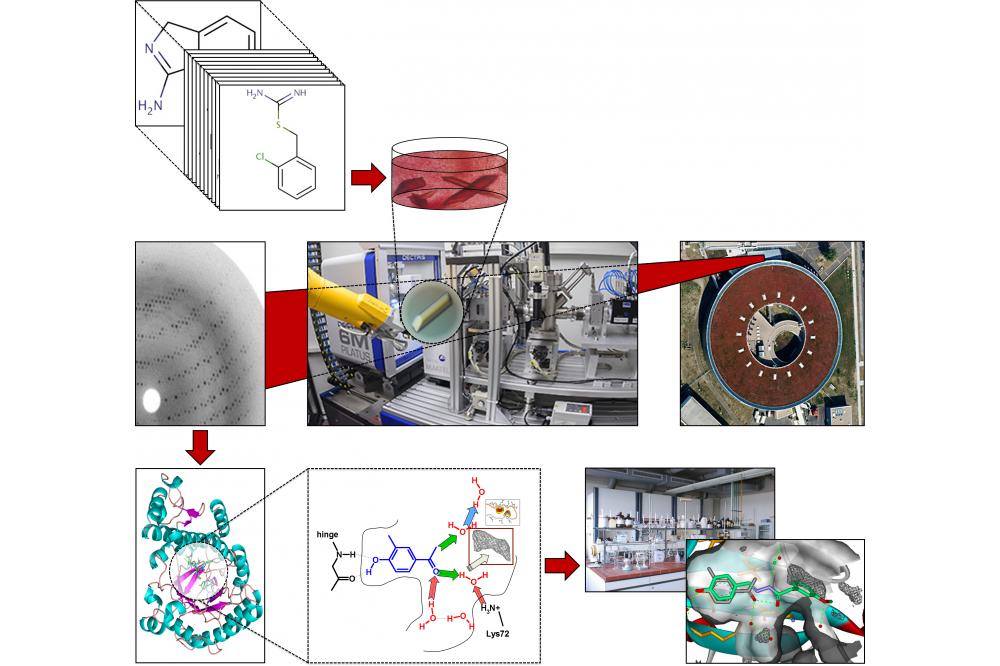
Ligand binding pockets differ regarding their shape and arrangement of potential interaction sites. Thus, we design a general-purpose 3D-diverse fragment library for crystallographic fragment screening that can address a great variety of binding sites while providing particularly suitable starting points for subsequent drug discovery efforts. To facilitate fragment-to-lead evolution after hit identification, this library must adequately cover the accessible chemical space of fragments including substructures of drug-like ligands.
A reasonable coverage of chemical space aims at fully exploiting the fact that already a small selection of ~103 appropriate fragments covers a much larger proportion of the overall chemical fragment space (~107 compounds) than a typical high-throughput screening collection (105 – 106 compounds) with respect to the drug-sized chemical space (~1063 compounds, MW < 500 Da). Despite the relatively low affinity of fragments to their target, their high ligand efficiency (affinity divided by number of non-hydrogen atoms) makes them excellent starting points for drug discovery. In addition, fragments often bypass the strict steric requirements for the binding of larger drug-like ligands, thus leading to high hit rates up to 15%. This allows direct crystallographic screening by crystal soaking of a small and preferably target- or structure-based selection of 102 – 103 fragments.
Methods
To complement our established in-house 364-fragment library, [1] we now compile a set of 1000 high-quality fragments that are particularly suited for crystal soaking, immediate fragment-to-lead evolution, and prepared for computer-aided subset selection and lead design. This expanded library will be part of the Frag2Xtal service facility for crystallographic fragment screening, which will be available at semi-automated crystallographic beamline 14.2 at the BESSY II storage ring (Berlin). Aiming at a representative coverage of chemical space, all sufficiently available and reasonable fragments (> 250,000 compounds adopting > 1.4 • 106 conformational and molecular states) were clustered into groups of 3D-similar compounds.
Results
To this end, > 1012 pairwise molecular similarities were calculated as the 3D overlap of the volume and spatial distribution of interaction features (charges, hydrogen bond donors/acceptors, aromatic ring, etc.) using the program ROCS (rapid overlay of chemical structures). [2] Based on these similarities, a hierarchical clustering using the software SparseHC [3] was carried out at the Marburg HPC cluster MaRC2 taking advantage of its large memory capacity.
Outlook
Selecting favorable representative fragments from each cluster will allow covering the available chemical space with fragments that are particularly suited for crystallographic fragment screening and as starting points for drug discovery projects.