Optical Spectroscopy, Dielectric Properties and Structure Discrimination of SnN (20 ≤ N ≤ 40)
 Filip_Rivic_Optical Spectroscopy, Dielectric Properties and Structure Discrimination of SnN (20 ≤ N ≤ 40)_Figure1
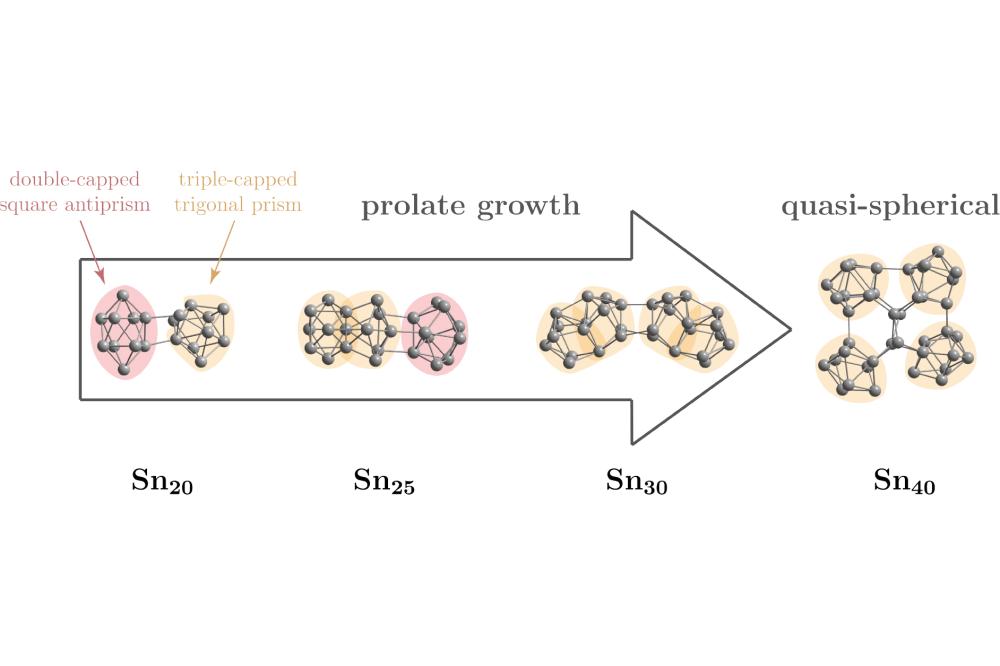
Filip_Rivic_Optical Spectroscopy, Dielectric Properties and Structure Discrimination of SnN (20 ≤ N ≤ 40)_Figure1Figure 1: Schematic display of the prolate cluster growth to a quasi spherical structure for the predicted ground state structures (GM) for SnN with N = 20; 25; 30 and 40 at the PBE0/def2-TZVPP level of theory. Tin atoms are displayed as gray spheres and the two building blocks are highlighted in red (double-capped square antiprism) and yellow (triple-capped trigonal prism).
Introduction
The previous investigation of neutral SnN clusters with N = 6 - 20 showed that for clusters with N ≤ 15 prolate structures are dominant. However, it is predicted that for larger tin clusters quasi-spherical structures occur again. Therefore, structures were calculated the geometric and electronic structures and the transition of SnN clusters in the range of 20 ≤ N ≤ 40 from prolate to spherical geometries was investigated. The calculated geometric structures can then be compared to experimental data gained in our accompanying electric beam deflection and photodissociation spectroscopy experiments by simulating the rotation in the electric field or electron dynamics, respectively. With this, it is possible to discriminate and verify the theoretically suggested structures.
Methods
Energetically-favored initial geometries were generated using a plane-wave Density Functional Theory-based (pw-DFT) Genetic Algorithm (GA) developed in our group. These calculations are performed using the Quantum Espresso software package. From the generated pool of structures candidates in the range of 2 eV relative to the energy of the energetically lowest-lying isomer are optimized on the LC-ωPBEh/def2-TZVPP and PBE0/cc-pVTZ-PP level of theory and verified by frequency analyses for the ground state. The absorption spectra are computed via Time-Dependent DFT (TDDFT) and are further analyzed by a molecular orbital and natural transition orbital analysis making extensive use of symmetry considerations. Additionally, CCSD(T)/cc-pVTZ-PP single-point energies on top of the PBE0/cc-pVTZ-PP optimized geometries have been computed using Orca v5.0.1.
Results
The geometric structures and absorption spectra of SnN (20 ≤ N ≤ 40) were calculated within this project. For the studied size range, triple-capped trigonal prisms and doublecapped square antiprisms were found to be the most prominent building blocks for larger tin cluster assemblies. Additionally, it can be observed that a transition from prolate to quasispherical geometric structures occur in the size range 30 ≤ N < 40. By comparing the results from the quantum chemical calculations with the experimental data, we found that the prolate isomers are predominantly present in the molecular beam for all but the Sn40 cluster which adopts a quasispherical geometric structure
Discussion
Even though the structural transition from prolate to compact structures is well known for semiconductor clusters, its exact transition point is still under debate for neutral tin clusters. By comparing the data calculated within this project with the experiment, we now further narrowed down the size range of the transition. In comparison to previously investigated cationic tin clusters of this size regime, very similar geometries were obtained that appear to be slightly less distorted according to their closed-shell spin configurations. In the future it would be interesting to extent the size range even further to also account for the structural transition towards the respective bulk modification.