Investigation of the Electronic Structure of Nitrogen Photoactivation Complexes
 Severine Rupp_Investigation of the Electronic Structure of Nitrogen Photoactivation Investigation of the Electronic Structure of Nitrogen Photoactivation Complexes_Figure1
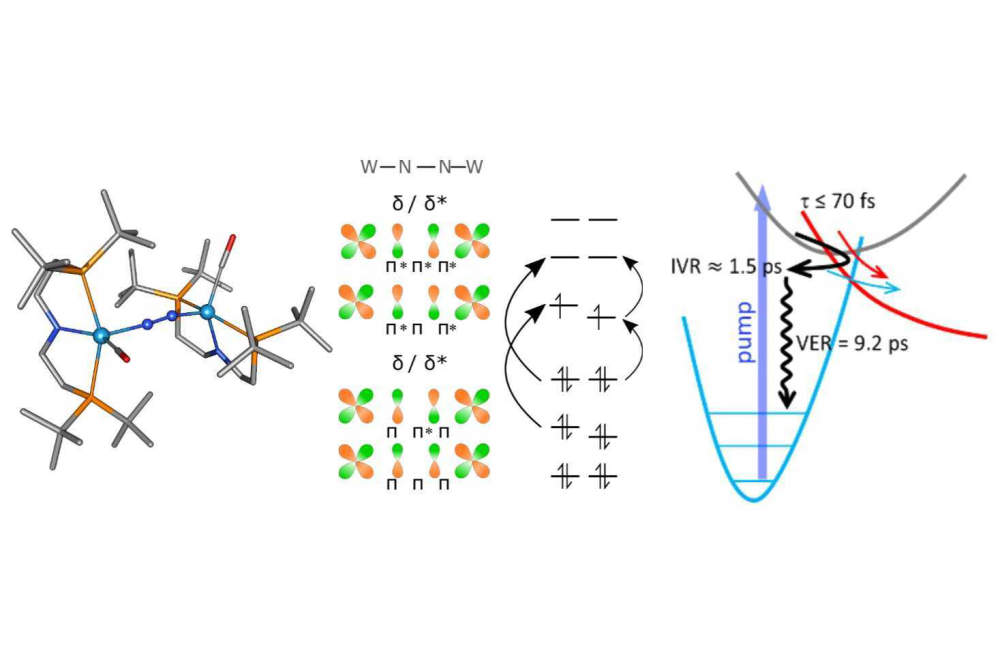
Severine Rupp_Investigation of the Electronic Structure of Nitrogen Photoactivation Investigation of the Electronic Structure of Nitrogen Photoactivation Complexes_Figure1Figure 1: The left panel shows the nitrogen-activating tungsten complex of Schneider and co-workers. The middle panel illustrates its electronic structure with a molecular orbital scheme and exemplary electronic excitations. We identified the following possible photochemical processes as shown in the right panel: a direct dissociation (red arrow) and a quasi-thermal N-N photodissociation (black/blue arrows; adapted from JACS Au 2021, 1, 6, 879–894).
Severine Rupp
Introduction
Ammonia derived from atmospheric N2 and fossil H2 in the Haber-Bosch process represents the main source of nitrogen compounds as a base chemical for the synthesis of value-added products. In order to access a more diverse range of N-containing chemicals it is of high interest to develop tailored nitrogen-activating catalysts. One way of achieving synthetic nitrogen fixation is selective light-driven N2 cleavage. What is currently known is that all of the complexes capable of nitrogen photoactivation have a linear core of the form M-N-N-M which undergoes a geometric and electronic change during the light excitation process. It is the aim of this project to describe the electronic structure of such complexes using a high level of theory to gain a better understanding of the photophysical and photochemical processes involved in nitrogen activation. Specifically, we aim to observe the electronic structure changes involved in the cleavage of the formal triple-bond of dinitrogen by characterising the excited state manifold of our systems.
Methods
This project mainly uses density functional theory (DFT) and time-dependent density functional theory (TDDFT) to evaluate the electronic structures of nitrogen activating transition metal complexes and to support the interpretation of the experimental results provided by collaborators. As analysis tools we use the computed electronic excitation and vibrational spectra, molecular orbitals, difference densities and the transition analysis tools of the TheoDORE programme.
Results
We identified characteristic electronic features of nitrogen activating complexes using the molybdenum and tungsten complexes synthesised by Sita and co-workers as an example. Using the TheoDore analysis we could differentiate between thermal and photoactivating complexes by characterising the electronic transition character. We identified that significant ligand-to-metal-charge transfer character is a requirement for a potentially photoactivating electronic transition. For the tungsten complex designed by Schneider and co-workers, we described its thermal and photochemical dinitrogen splitting reactivity. We reproduced the experimental thermochemical data computationally learning that the conformation of the rather flexible pincer backbone plays a crucial role in the reactivity of tungsten complex. We investigated the photochemical processes computationally, considering electronic and vibrational excitations. In conjunction with experimental transient absorption and resonance Raman data, we were able to identify the photochemical process of a quasi thermal N-N photodissociation. Future studies will aim at distinguishing between excited state reactivity and hot ground state reactivity, which will require a higher time resolution.
Discussion
Future work will focus on analysing the photochemical nitrogen activation of rhenium complexes by Schneider and co-workers on the TDDFT level scanning along the N-N bond length as designated reaction coordinate. Furthermore, a more precise description of the electronic structures using a higher level of theory such as CASSCF/CASPT2 is targeted to overcome the intrinsic limitations of the DFT/TDDFT approach.