Quantifying the Dynamic Acceleration upon Coarse-Graining Using Machine Learning
 Saientan Bag_Quantifying the Dynamic Acceleration upon Coarse-Graining Using Machine Learning_Figure1
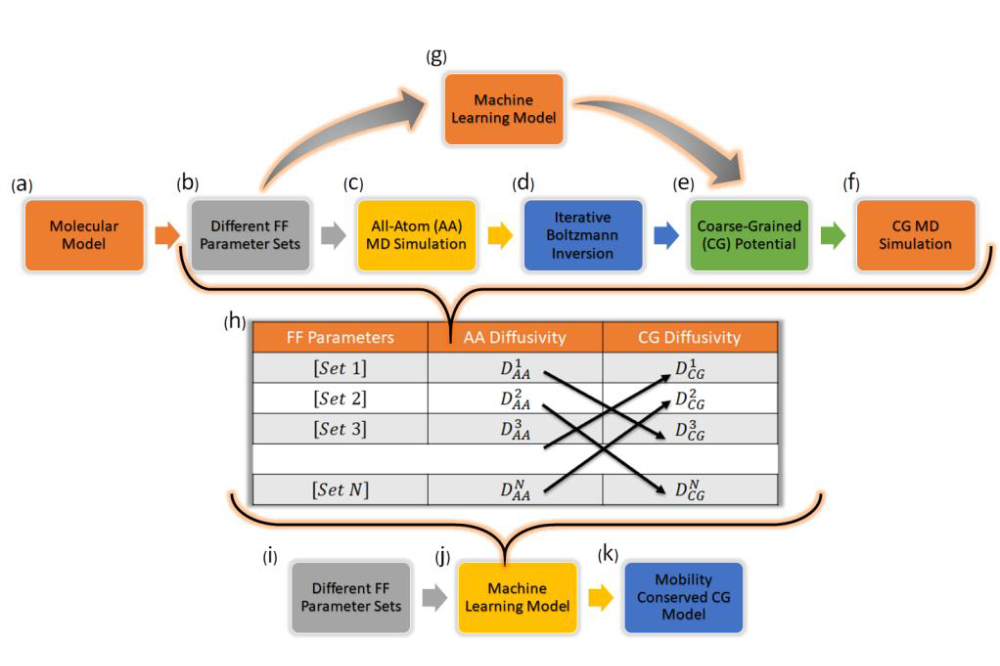
Saientan Bag_Quantifying the Dynamic Acceleration upon Coarse-Graining Using Machine Learning_Figure1Figure 1: Workflow to design machine learning models to predict a simulation-ready mobility-preserving coarse-grained model (for molecular dynamics simulation) starting from an all-atom model.
Saientan BagIntroduction
Coarse-Grained molecular dynamics simulation is a promising alternative to all-atom molecular dynamics simulation for fast calculation of system properties which is imperative in designing materials with specific target properties. There have been several coarse-graining strategies developed over the past few years which provide reasonably accurate structural properties of the system. But these coarse-grained models share a major drawback by introducing an artificial acceleration in molecular mobility. In this context, we asked if we could make the coarse-grained models more accurate. With this question in mind, in this project we aimed to design machine learning models to ensure more accurate prediction of the system dynamical properties from the coarse-grained molecular dynamics simulation. We successfully designed a machine learning model in the form of an artificial neural network which directly predicts the simulation ready mobility preserving coarse- grained potential as an output given the all-atom force-field parameters as input. To generate the training data for the machine learning model we had to perform approximately 300 sets of all-atom and coarse-grained simulation which was performed on Lichtenberg Hochleistungsrechner.
Methods
We constructed a workflow to develop machine learning models for structural and dynamical property preserving coarse-grained potential for a molecule. We presented a “proof of concept” demonstration of the workflow by choosing 2,3,4 trimethylpentane as a model system. We first performed all-atom molecular dynamics simulations for many different random choices of the bonded force-field parameters of 2,3,4-trimethylpentane. We further performed Iterative Boltzmann Inversion to generate coarse-grained potential corresponding to each all-atom molecular dynamics run. Coarse-Grained molecular dynamics simulations are performed in the end with the Iterative Boltzmann Inversion generated coarse-grained potential. A large set of all-atom and coarse-grained simulation runs are performed and machine learning models are trained based on the data obtained from these runs. We trained a neural network which directly learns the Iterative Boltzmann Inversion generated coarse-grained potential and therefore this is structural property-preserving. To build the data set (for machine learning model) for a dynamics-preserving coarse-grained model we look for pairs of systems such that the all-atom diffusion coefficient of one system becomes equal to the coarse-grained diffusion coefficient of the other. Another neural network was trained based on this dataset to learn the relation between the all-atom force-field parameters of one system and coarse grained potential of the other system. Therefore these coarse-grained models are dynamical property-preserving.
Results
We have developed different machine learning models with the general goal of obtaining correct dynamical properties from the coarse-grained simulation. We demonstrated how a ML model can be constructed that directly predicts the simulation-ready coarse-grained potentials which preserve either structural or dynamical properties with only the all-atom force field parameters as input. So far, the machine learning model generated coarse-grained potential cannot reproduce the structure and dynamics of an all-atom model simultaneously equally well. Therefore, the strategy of generating a structure-preserving coarse-grained model by iterative Boltzmann inversion or machine learning first and then rescaling its dynamics using a machine learning predicted acceleration factor might be the method of choice for the time being. The applicability of this machine learning model has only been demonstrated for a specific molecule, namely 234TriMePe. A natural question arises at this point is what about the transferability of this ML model to other molecules? In the future we plan to generalize our machine learning models to include other molecules by generalizing the input features.
Discussion
In the last decades an enormous effort has been made to design new algorithms and software for user-friendly and efficient molecular dynamics simulation. This resulted in a massive increase in the use of the molecular dynamics simulation in various research fields. But the all-atom molecular dynamic simulations are still computationally expensive to perform and it requires a computer cluster for real-life systems. What does it take to make molecular dynamics simulation doable on a desk-top computer? Coarse-Grained molecular dynamics simulations seem to be a possible solution. However, coarse-grained molecular dynamics simulation has yet to reach the accuracy of the all-atom molecular dynamics simulations. The current project is the result of our continuous effort to make coarse-grained simulations more accurate in-terms of its capability to predict better dynamical properties of the systems. We believe in the marathon of designing accurate coarse-grained potentials, our data driven approach is a step forward.