Understanding and Exploiting an Iron Catalysed Route to New Chemical Synthons
 Martin_Diefenbach_Understanding and exploiting an iron catalysed route to new chemical synthons_Figure1
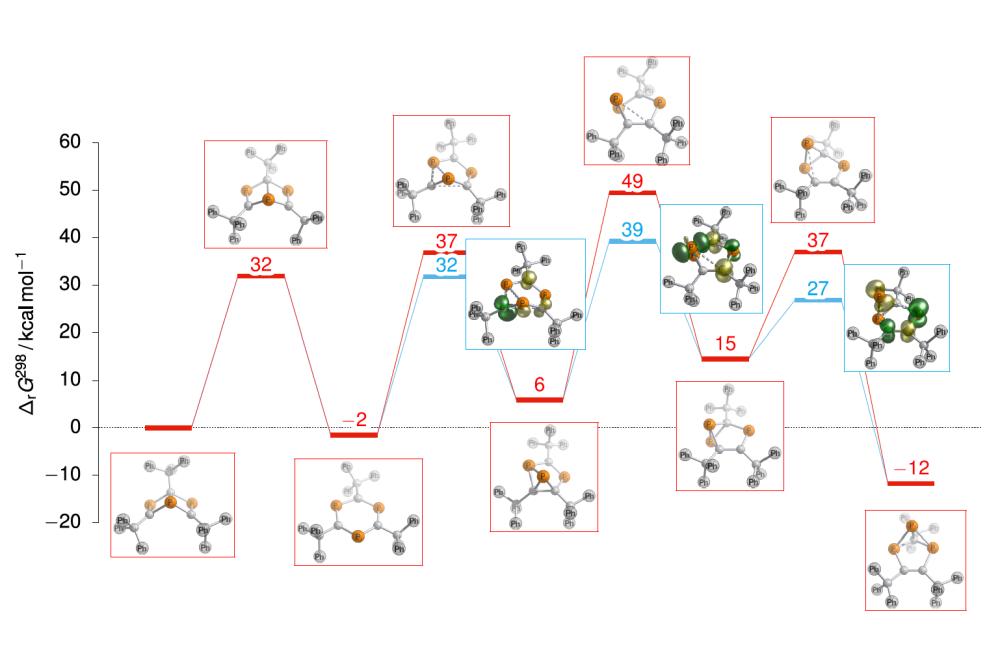
Martin_Diefenbach_Understanding and exploiting an iron catalysed route to new chemical synthons_Figure1Figure 1: Computed rearrangement of Dewar phosphabenzene towards novel triphospha(tricyclo) species. Energies for closed-shell singlet species in red colour and for broken-symmetry species in light blue colour. Spin densities of broken-symmetry transition structures illustrate radical character.
Martin DiefenbachIntroduction
The project focuses on an iron catalysed method to prepare unprecedented chemical synthons, developed in the group of Dr. Webster (University of Bath). The aim is to explore the catalytic
cycle with computational means by the use of quantum chemical calculations. This involves both, the identification of key intermediates and transition structures en route to product formation, as well as investigation of potential catalytically active transition-metal species itself. In the experimental lab, an iron-catalysed route to produce the elusive Dewar phosphabenzene on a gram-scale was recently established, and its subsequent rearrangement to a novel triphospha(tricyclo) species, as well as the onward coordination chemistry was experimentally demonstrated. Quantum chemical investigation of the Dewar phosphabenzene and its rearrangement products are carried out to unravel the underlying mechanism.
Methods
The study of Dewar phosphabenzene and its thermochemical and kinetic stability with respect to isomerisation, rearrangement and coordination chemistry, involves mostly main-group element chemistry. The computational investigation of mechanistic details was performed using density functional theory (DFT). For benchmarking of the DFT results and for further scrutinising the results of critical species, which also include iron(II) complexes, validation of these methods through correlated coupled cluster theory was applied.
Results
DFT calculations provide a four-step mechanism for the rearrangement of Dewar phosphabenzene to the novel triphospha(tricyclo) species found in the experimental study. The predicted mechanism involves open-shell transition structures with radical character, which is consistent with the experimental information. Calculations of corresponding Au complexes with Dewar phosphabenzene and its rearrangement isomers predicted a rich coordination chemistry potential, which could be confirmed expermimentally by NMR spectroscopy and X-ray crystallography.
Discussion
The quantum-chemically predicted mechanism for the rearrangement of Dewar phosphabenzene to yield the novel triphospha(tricyclo) species proceeds via a six-membered triphosphabenzene ring structure and subsequent ring-contraction involving concerted P–P and C=C bond formations. The barriers for the rearrangement are in accordance with experimental observations only, if radical character is accounted for at the sites where bonds are formed or broken (Figure 1). The coordination chemistry potential of the triphospha-heterocyclic isomers was demonstrated by computational prediction and experimental formation of gold complexes. Where X-ray crystal refraction data were not available, DFT calculations could further substantiate the site of coordination obtained from NMR spectroscopic data. First insight into potential catalytically active iron-salen complexes was obtained at a systematically convergent high-level ab initio coupled-cluster approach. Prediction of the electronic ground state multiplicities is of fundamental importance, and the high-spin state is found to be the most stable in the iron complexes studied thus far.