Electronic Structure Calculations on the Influence of Alkali Metals on the Properties of Ag-Containing Cu(In, Ga)(S, Se)2 Absorber Materials
 Markus_Mock_Electronic Structure Calculations on the Influence of Alkali Metals on the Properties of Ag-Containing Cu(In, Ga)(S, Se)2 Absorber Materials_Figure1
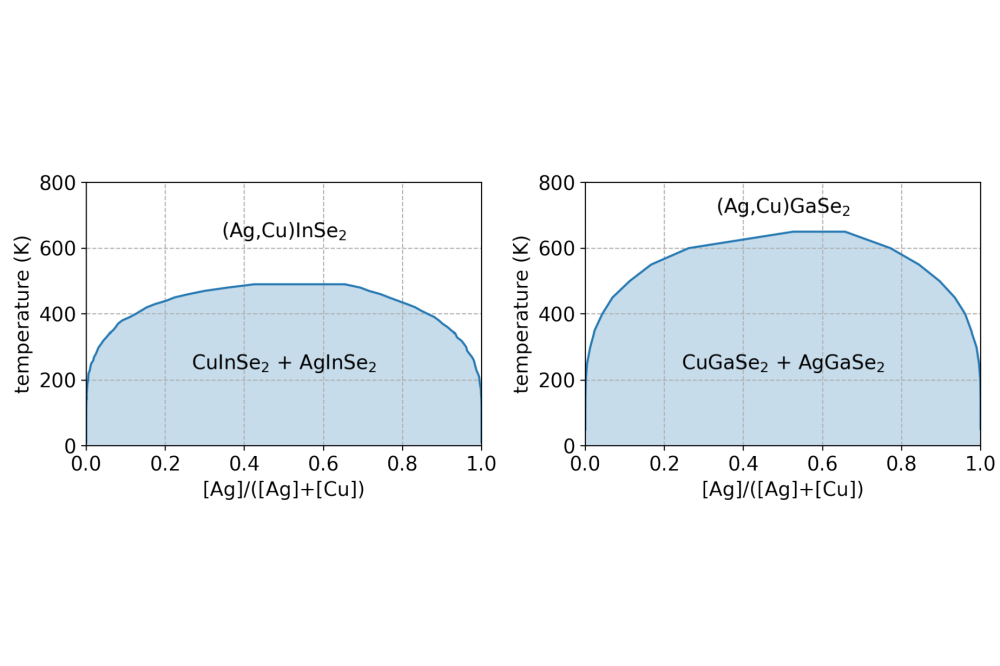
Markus_Mock_Electronic Structure Calculations on the Influence of Alkali Metals on the Properties of Ag-Containing Cu(In, Ga)(S, Se)2 Absorber Materials_Figure1Figure 1: The phase diagrams of (Ag, Cu)InSe2 and (Ag, Cu)GaSe2 calculated using semi-grand canonical Monte Carlo simulations.
Markus MockIntroduction
The compound semiconductor system Cu(In, Ga)(S, Se)2 (short: CIGS) forms the basis for the currently most efficient thin-film solar cells with record laboratory efficiencies in the range of 22% to 23%. In general, Ga and S are used to optimize the band gap in the absorber. An alternative way of increasing the band gap is to substitute Ag for Cu. Ag-containing CIGSe cells that were produced by co-evaporation were able to achieve efficiencies of up to 21% under laboratory conditions. The main goal of this project is the examination of the influence of Cu substitution by Ag on the electronic and structural properties of CIGS solar cells absorber materials. For this objective density-functional theory (DFT) calculations with the VASP software will be used to parameterize a cluster expansion model. The cluster expansion model can then be used in semi-grand canonical Monte Carlo simulations to investigate the phase diagram of the system. The parameterization of the cluster expansion model requires an extensive set of training structures to learn the relevant interactions within the system.
Methods
In this project we employed density-functional theory (DFT) calculations to train a cluster expansion model. These calculations were done using the software package VASP, which is already successfully in use on the Lichtenberg cluster as well as in our group and has proven to be a reliable and high-performance code. The training set calculated using VASP is going to be used to train a cluster expansion model using the ICET software package (https://icet.materialsmodeling.org/index.html).
Results
A cluster expansion (CE) model was constructed based on a database of 242 training structures in the (Ag, Cu)InSe2 case and 108 training structures in the (Ag, Cu)GaSe2 case. The training structures were generated by enumeration of possible arrangements of Ag and Cu on the (Ag, Cu)(In/Ga)Se2 lattice. The software package ICET was used to fit the cluster expansions to this database. These cluster expansions were used in the following sections to perform semi-grand canonical Monte Carlo simulations to investigate the phase diagram of the system. Figure 1 finally shows the comparison between the phase diagrams of (Ag, Cu)InSe2 and (Ag, Cu)GaSe2. Both show a similar behaviour with a solid solution at elevated temperatures and phase seperation at low temperatures. The upper consolute temperature of 650 K is higher for (Ag, Cu)GaSe2 compared to the 490 K for (Ag, Cu)InSe2.
Discussion
In this work we examined the influence of Cu substitution by Ag on the electronic and structural properties of CIGS solar cells absorber materials. We parameterized a cluster expansion model of the (Ag, Cu)InSe2 and of the (Ag, Cu)GaSe2 system based on density-functional theory calculations. The cluster expansion model was then be used in semi-grand canonical Monte Carlo simulations to investigate the phase diagrams of both systems, showing that the system has a tendency to phase separate at low temperatures, while it shows a solid solution at high temperatures. The transition temperature between these states is significantly higher for (Ag, Cu)GaSe2 compared to (Ag, Cu)InSe2.