Ab-initio Modeling of Battery Materials
 Sadowski, Marcel_Ab-initio Modeling of Battery Materials
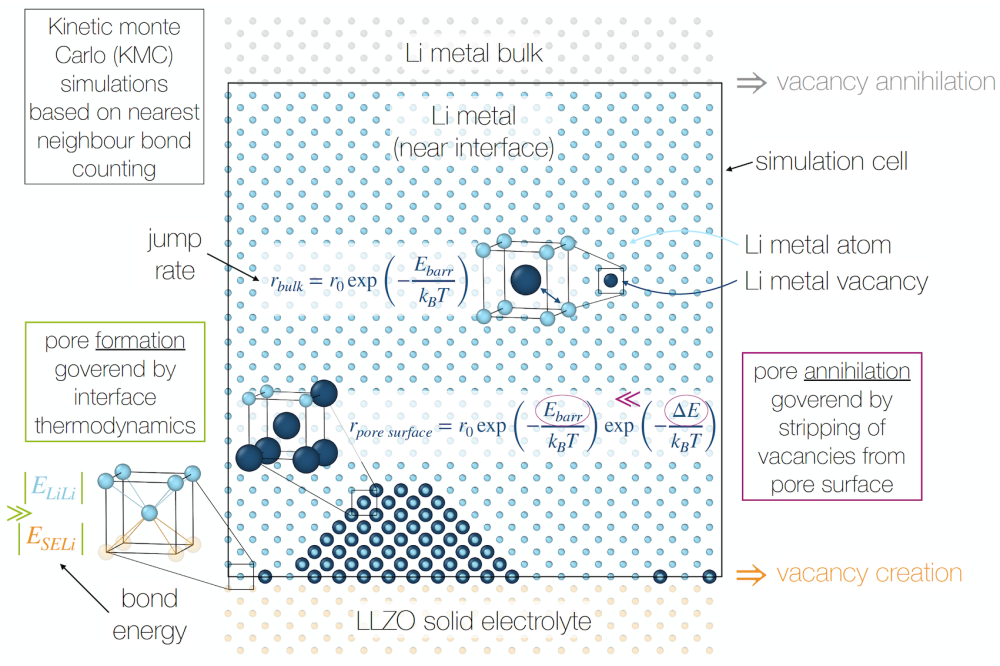
Sadowski, Marcel_Ab-initio Modeling of Battery MaterialsFigure 1: Modelling approach to investigate the pore formation kinetics at interfaces between solid electrolytes and lithium metal anodes.
Marcel SadowskiIntroduction
One approach to the realization of safer batteries relies on all solid-state batteries (ASSB) which use a non-flammable solid electrolyte (SE) instead of the commercial flammable liquid organic electrolytes. While many obstacles to the successful production of these batteries have already been overcome, the inner and outer interfaces in a real battery setup remain a major challenge. Thus, a thorough understanding of the interfacial atomistic processes is crucial, highlighting the value of interface simulations on the atomic scale. Currently, these are only possible via ab-initio methods, such as density functional theory (DFT) calculations, because no classical interatomic potentials exist, which can simultaneously describe the SE and electrode materials. As DFT is a comparatively expensive method, HPC facilities are necessary to effectively execute such calculations, especially for interface setups which usually contain a large number of particles.
Methods
Ab initio calculations based on DFT have been carried out using the Vienna ab initio simulation package (VASP) with projector augmented-wave pseudopotentials and the PBE exchangecorrelation functional. For all investigated materials, calculation parameters like cutoff energy of the plane-waves and k-point density have been converged prior to the production steps. Initial atomic structures were taken from XRD measurements, either determined by our experimental collaborators or from literature. All structures containing partially occupied atomic positions, the supercell program was employed. Ab initio molecular dynamics (AIMD) was used to analyze the Li transport properties of bulk phases via the mean-square displacements (MSD) and across interfaces by a custom post-processing protocol. Local jump events were additionally inspected using the Nudged Elastic Band (NEB) method. The kinetic Monte Carlo (KMC) simulations based on a nearest neighbour bond counting were performed by a custom python code run on local machines. Input parameters, however, were obtained by DFT calculations performed on the Lichtenberg cluster. Graphical and data analysis were performed with the Open Visualization Tool (OVITO).
Results
Subproject 1 – Pore formation and evolution at the LLZO|lithium metal interface: Pore formation at the lithium metal anode (LMA)|SE interface is a key obstacle to utilising LMA in ASSB at the industrial level and oftentimes attributed to insufficient vacancy diffusion within the LMA. In order to computationally address the kinetics of this system at finite temperature, KMC simulations were performed. It was found that neither the formation, nor the annihilation of pores are a result of slow vacancy diffusion: While the former is mainly influenced by the interface thermodynamics between the LMA and the SE, the latter is limited by the stripping of vacancies from the pore surface.
Subproject 2 – Internal interfaces in the garnet LLZO: Since LLZO is usually synthesised in powder form, followed by a sintering step in order to achieve high densities, leading to poly-crystalline pellets, the percentage of grain boundaries (GB) within the SE itself is significant. In order to judge the influence of these GB on the Li diffusion, AIMD and NEB calculations of symmetric ∑5(210) tilt GBs were conducted. These revealed, that the Li mobility is significantly hindered by the GB, reducing the Li diffusivity by ≈ 50%.
Subproject 3 – Stability and conductivity of the LATP|NCM interface: Interfacial instability between the NASICON-type LATP (Li1.3Al0.3Ti1.7P3O12) SE and the NCM (LiNi0.6Co0.2Mn0.2O2) cathode still prevents successful application of this material combination in ASSB. Regardless of the chosen LATP/NCM surface termination, an interfacial reaction was observed during 30 ps long AIMD simulations above 650 °C, which is characterized by an interdiffusion of LATP and NCM particles. This result was later also obtained by Scanning Transmission Electron Microscope (STEM) imaging.
Subproject 4 – Interfaces involving lithium thiophosphate glass-ceramics: A set of interface structures has been generated and remained stable at moderate temperatures during the simulations. The presence of a glassy phase seems to not promote Li+ transport for crystalline phases with poor ionic conductivities. For systems with better transport properties, the statistics did not allow for a precise determination of interface resistances and the simulations suggest that the individual bulk contributions are most important for the overall properties of the glass-ceramic.
Subproject 5 – Internal interfaces in the argyrodites Li6PS5X (X=Cl, Br): Symmetrical ∑5(001) tilt and twist GBs with different degrees of Br-/S2- site-exchange for the Li6PS5Br system were constructed and subjected to AIMD simulations, which revealed the ambivalent character of GBs with respect to the Li+ transport properties. At 0% site-exchange GBs enhance the local Li+ diffusion because the well defined cage structure is distorted. At 25% and 50%, however, GBs slightly hinder the Li+ diffusion, presumably because diffusion pathways are less favorable across the interface compared to the bulk.
Discussion
Regarding the process of pore formation at the LMA, the factors responsible for vacancy agglomeration have been identified. A practical strategy for improving the SE|LMA contact, however, has not been defined yet, since an atomically flat surface without impurities is not feasible for industrial application. Thus, the effect of an ionic liquid interlayer will be investigated, which has experimentally been shown to improve the interfacial contact significantly.
With respect to the influence of GB on the Li mobility in SE, a systematic difference between sulfide and oxide type SE has been found: While diffusion in the former is only slightly affected by the presence of GB, the latter display a pronounced reduction of diffusivity due to GB. To ensure, that this behaviour is not particular for LLZO, the Na+ SE NASICON has been investigated as well, producing the same result. We expect that this reduction of mobility is due to a higher density within the GB with respect to the bulk phases. Thus, bottlenecks for ion transport, formed by the surrounding oxygen ions, are reduced. In contrary, for sulfide SE the density in the GB is not significantly affected. In order to further strengthen this hypothesis, additional calculations of grain boundary setups need to be done.
Finally, regarding the LATP|NCM interface simulations, the experimentally observed instability was reproduced by DFT calculations suggesting that it is an intrinsic feature of this material system and not a result of impurities/side phases present in experiment. Thus, possible protective coatings will be examined next.