Testing and Development of Methods for the Calculation of Phase Diagrams of Interatomic Potentials
 Helzel, Aron_Testing and Development of methods for the calculation of phase diagrams of interatomic potentials
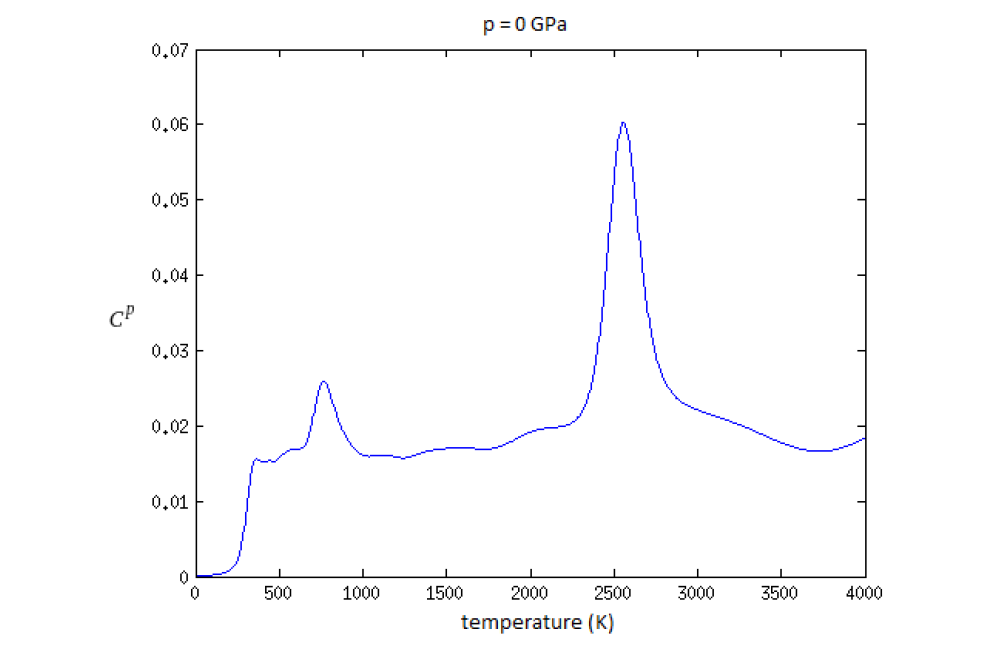
Helzel, Aron_Testing and Development of methods for the calculation of phase diagrams of interatomic potentialsFigure 1: Specific heat plotted against the temperature for the Müller-Albe iron potential with a Nested Sampling run with 60 atoms and 4800 walkers. The peaks resemble the transition points with the melting point at about 2500K and a solid-solid phase transition at about 800K. At about 300K the run was stopped, which causes the calculated specific heat to drop to zero at this point.
Aron HelzelIntroduction
Understanding the behavior of different materials not only furthers general knowledge, but can also often be used for practical purposes in e.g. engineering. Computer Simulations, where these materials are simulated as small atoms, that exert forces on each other, play an important role in this field and there are already many applications for many different materials. Since these simulations cannot describe the behavior of the materials without approximations and simplifications, it is always important to assess their error and compare results with experiments, where this is possible. One of the properties, that can be compared, are phase transitions, which are changes in the structure of the material, that happen at certain temperatures, pressures and compositions of the material. In this project one of the methods for calculating transition points (Nested Sampling) was investigated on its strengths and weaknesses. There was a strong focus in the improvement in the usage of this method especially for curbing the errors that appeared. For this many test calculations were necessary. In the end a systematic search for the transition temperatures at different pressures for iron with one specific potential, from which the forces are calculated (Müller-Albe bond-order potential), was done, that was more accurate than calculations that were done, before the improvements were made. Since such calculations are quiet costly, it was necessary to use a High Performance Computer. Furthermore for a variety of other materials (Ga, As, GaAs and GaN) the method was further assessed in its ability to predict the best arrangement the atoms can take in space using potentials specific to these materials. Finally another method (USPEX), that can be used for these purposes, was started to be investigated on for comparison.
Methods
The Nested Sampling method was published in the open source software pymatnest. It starts with a sample of atom configurations, where the atoms are placed at random in a space of high volume, which resembles the structure of a gas. During the run of the program the configurations, that are connected with the highest temperature, are culled from the set and replaced with configurations of lower temperature. In this way configurations of lower and lower temperatures are generated. Also the speed, in that the temperature gets lower with each step of the algorithm, is connected to the specific heat of the material, which is especially high at transition temperatures. In the end plotting the specific heat against the temperature can show all transition temperatures and the calculated configurations can be used to classify the phases (see Figure 1). The USPEX method was also published as open source software. It starts with a sample of atom configurations, that are highly symmetrical and resemble typical configurations for solid materials. The configurations are then varied through a random choice of certain varying operations, that are mostly preset, but can also be modified. Through this a number of good configurations for the given potential can be found. The transition temperatures between them can then be found through further calculations.
Results
With the tests on the iron potential it could be found, that the Nested Sampling method produces significant errors in the transition temperatures. At least for melting temperatures it is possible to get rid of most of the error through the use of many more atoms in the simulations. For other transition temperatures this is not possible, since the computing time would get too high. Borrowing from a mathematical variant of the Nested Sampling method some optimisations could be made in hopes of lowering the computation times again. As a result more atoms could be used than before, so that the error became smaller. Since the parameters can be set in such a way, that the calculation times get smaller, but the chance for not reaching the correct configurations increases, another idea was the use of many shorter calculations, of which most fail, but where the most successful ones can be picked and analyzed. This actually achieved good results in some cases, but for high numbers of atoms this was not the case anymore. Using this knowledge the transition temperatures for the iron potential under a variety of pressures could be calculated. Furthermore at least for GaAs the method found the correct zero temperature configuration. This was not the case for the three other materials.
Discussion
With this work the strengths and weaknesses of the Nested Sampling method could be analyzed and the usage of the method could be improved. Although computation costs and errors of the method are still quite high, the method still has some advantages in comparison to other methods, such as the lack of prior knowledge about the potential. Nevertheless USPEX is a promising method, that also possesses some of the advantages of Nested Sampling and has much lower computational cost. Testing of this method already started and will be done in the coming months.