Molecular Dynamics Simulation of a Tribology System
 Mehrnia, Seyedmajid_Molecular dynamics simulation of a tribology system
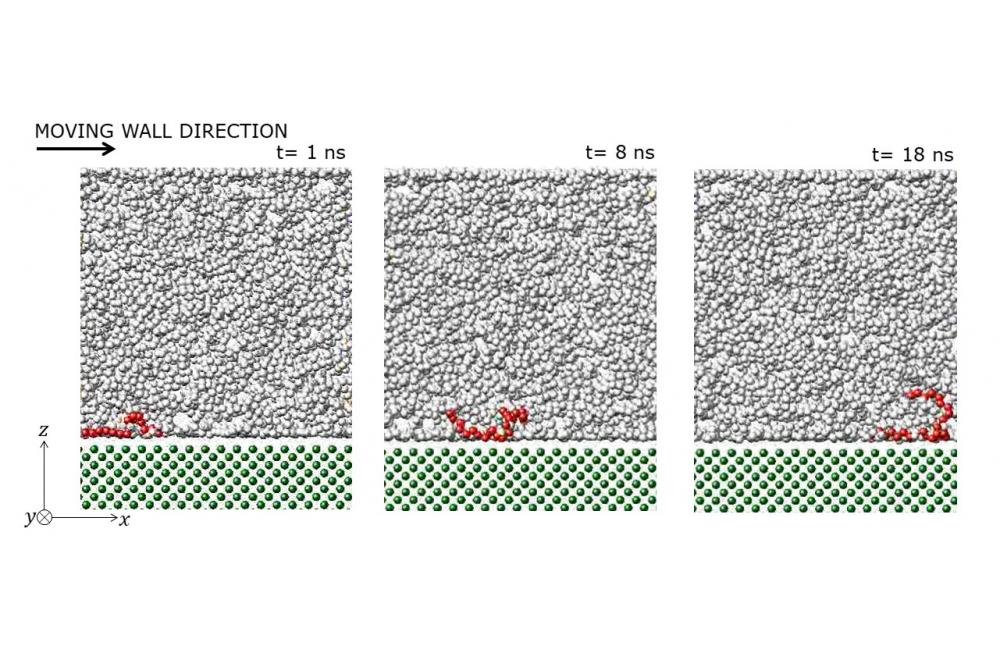
Mehrnia, Seyedmajid_Molecular dynamics simulation of a tribology systemFigure 1: Side views of 1-Decane tetramer simulation box for tracking a molecule adjacent to the stationary surface.
Seyedmajid MehrniaIntroduction
The MD method is suitable for studying the behavior of liquid molecular systems. According to past investigations in the modeling of hydrocarbon oils in tribology systems, a study of the slip length of PAO oils confined between iron surfaces has not been accomplished. Therefore, we employed nonequilibrium MD modeling of a shear flow to study the wall slip, emphasizing the effect of the liquid molecule structures in interface with iron atoms surface. This project is aimed first to investigate the accuracy of the MD method in a shear flow, and second to investigate the slip length of the branched alkane oils in different temperatures with iron surfaces.
Methods
All models were designed using Avogadro software, then assembled and optimized by the Packmol software. The models utilized for the liquid molecules were a full atom style model that includes bond stretching, bending, and torsion. The MD equations of motion integrated using the velocity-Verlet algorithm with an integration time-step of 1.0 fs for the selected force-field. The MD modeling in this research was performed by the LAMMPS molecular dynamics simulator, an open-source code. Through all simulations, the temperature is uniformly controlled by a Langevin thermostat. This thermostat simulates a conductive heat flux through the surfaces and guarantees heat removal from the liquid.
Results
The results show that the slip length depends on the number of branches of the molecule structure. It observed that the velocity profiles varied at different temperatures, such that the slip velocity decreased by about 1.25% for a one-degree Celsius increased in temperature. The further analysis reported that in attendance of slip length, the PAO oil still gives an Arrhenius viscosity dependence on temperature. However, the liquid activation energy declines by decreasing carbon number and molecule branches, showing PAO fluidity due to the sliding phenomenon.
Discussion
It was tried to compare the results of this simulation to the experimental measurement and explaining some reasons for the differences. Undoubtedly, the assessed slip length is associated with an error margin in this project. Nevertheless, the wall sliding trend is consistent with the experimental results, which confirm our MD simulation result. It shall also notify it, increasing the oil film thickness expects an increase in the computational cost, so the discussion has been restricted in this MD simulation.