Ab-Initio Thermodynamics of Refractory Mo-Si-Ti Phases at High Temperatures
 Ab-initio thermodynamics of refractory Mo-Si-Ti phases at high temperatures
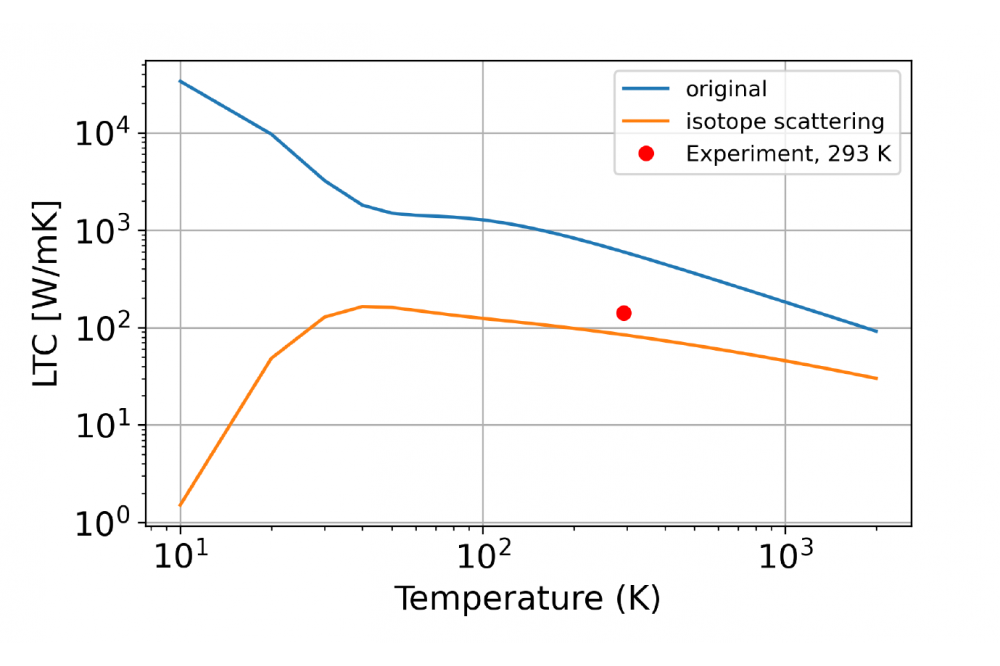
Ab-initio thermodynamics of refractory Mo-Si-Ti phases at high temperaturesFigure 1: Temperature dependent lattice thermal conductivity (LTC) of bcc molybdenum. The blue graph shows the LTC and diverges for T !0 since only phonon-phonon interactions are considered. The yellow graph shows the LTC when isotope scattering is introduced. The red dot shows the experimental value at 293 K.
David KasdorfIntroduction
Refractory metal silicide systems like Mo-Si-Ti are expected to be ideal substrates in composite materials for high temperature applications due to their deformability and toughness at ambient temperatures as well as extraordinary microstructural stability and creep resistance at high temperatures. Another crucial property is the tunable coefficient of thermal expansion which needs to be adjusted to the top layer with the purpose of keeping thermomechanical stresses in the multi-layered system as low as possible.
Typically, harmonic calculations based on density functional theory are used to calculate phonon spectra and corresponding properties. Anharmonic effects are then included with the help of perturbation theory. However, at the sought for high operating temperatures anharmonic effects become prevalent and lead to a breakdown of perturbation theory.
For this reason we employ the self-consistent phonon (SCPH) method to non-perturbatively compute phonons and anharmonic properties of Mo-Si-Ti phases. This includes the derivation of free energies, specific heats, thermal expansion coefficients and temperature dependent elastic properties. Moreover, we use the relaxation-time approximation within the Boltzmann transport equation (BTE-RTA) to calculate thermal conductivities including thermal anisotropies.
Methods
The Vienna ab initio simulation package (VASP) has been used to carry out calculations based on density functional theory (DFT). We applied projector augmented-wave pseudopotentials and PBE exchange-correlation functionals to all of our calculations. Calculation parameters like the plane wave energy cutoff and the k-point density have been carefully converged in advance of the production runs. The atomic structures were initially taken from the materials project database and subsequently optimized by an equation of state fit to the Birch-Murnaghan equation.
The generation of displacement patterns, fitting of force constants, BTE-RTA and SCPH calculations were governed with the help of the ALAMODE software package. The force calculations in the testing phase of the ALAMODE software package were conducted with the classical molecular dynamics code Large-scale Atomic / Molecular Massively Parallel Simulator (LAMMPS).
Results
In this project period the focus was to explore the the calculation of properties based on anharmonic lattice dynamics with help of the software package ALAMODE. At first we tested the program with force calculation based on interatomic potentials and the LAMMPS code. We learned that the symmetry of the cell and choosing the cutoff for interactions are crucial to find an acceptable amount of force calculations for a good accuracy of the lattice dynamical properties. In order to evaluate the lattice dynamics with forces from DFT calculations we converged the forces and optimized the cells of relevant Mo-Si-Ti phases, namely Mo, MoSi2, Mo3Si, Mo5Si3 and Ti5Si3.
We calculated force constants of third order for these materials. With these force constants and the Boltzmann transport equation in the relaxation-time approximation we calculated the lattice thermal conductivity (LTC). These LTCs diverge for T → 0 since only the scattering with other phonons limits their lifetime. Only when other scattering options are applied to the calculation, the LTC does not diverge. In the attached Figure we also show the LTC of molybdenum when isotope scattering is considered. In this case the phonons also scatter at the different isotope masses of molybdenum which limits their lifetime and the LTC accordingly.
However, it needs to be considered that the LTC is still modeled after a single crystal with no interfaces or grain boundaries. Those boundaries would further reduce the LTC which on the other hand increases the discrepancy with the experiment. Nevertheless, this discrepancy is easily explained with the metallic nature of molybdenum and the corresponding electronic contribution to the thermal conductivity.
Outlook
Currently, we are able to describe the lattice thermal conductivity of the Mo-Si-Ti bulk phases. In a real system however, interfaces and grain boundaries play an important role. In order to investigate the interfaces in an alloy like Mo-Si-Ti, the microstructure needs to be known. With the help of the above mentioned SCPH calculations we will be able to derive free energies of the involved phases. These free energies will be used to parameterize CALPHAD and phasefield models, which allow to determine the equilibrium microstructure of Mo-Si-Ti alloys with different compositions.
Also, the interfaces to top coat materials play a detrimental role. Only when those interfaces are strong enough the composite material will be able to withstand the thermomechnanical stresses at high temperatures.
For the next project period we plan to shift our focus to grain boundaries and interfaces while continuing with the calculation of bulk properties.