CCDCGAN: Inverse design of crystal structures
 Study for external electric field tuned thermal transport properties of two-dimensional materials
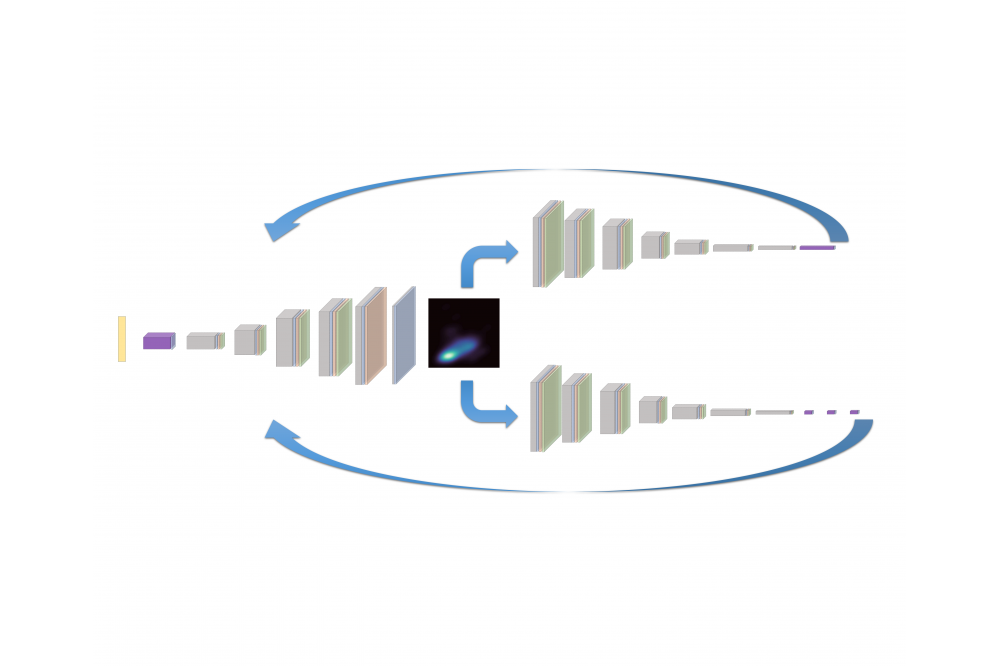
Study for external electric field tuned thermal transport properties of two-dimensional materialsFig. 1: Workflow of CCDCGAN.
Chen ShenIntroduction
Autonomous materials discovery with desired properties is one of the ultimate goals for modern materials science, and the current studies have been focusing mostly on high-throughput screening based on density functional theory calculations and forward modelling of physical properties using machine learning. Applying the deep learning techniques, we have developed a generative model which can predict distinct stable crystal structures by optimizing the formation energy in the latent space. It is demonstrated that the optimization of physical properties can be integrated into the generative model as on-top screening or backwards propagator, both with their own advantages.
Applying the generative models on the binary Bi-Se system reveals that distinct crystal structures can be obtained covering the whole composition range, and the phases on the convex hull can be reproduced after the generated structures are fully relaxed to the equilibrium. And by learning from the Materials Project database, the method generates a wide range of thermodynamically stable structures that do not exist in the database, covering intermetallic compounds, oxides, Li-containing compounds and chalcogenides, etc.
It is further demonstrated that increasing the number of structures learnt, or adding constrained models, can further improve the performance of the model. Thus, this model can pave the way to achieve the inverse design of materials with optimal properties.
Methods
The data to train this model has two different sources: first, by substitution from 10000 V-Ostructures and the followed DFT calculation, including Bi-Se, Co-Hf, etc.; second, direct down-load from Materials Project (MP) database. Then we transformed the known crystal structure into a valid representation, the crystal graph, via voxel model and auto encoder.
The reproduction ratio for specific system and multicomponent systems are 96% and 87%, respectively. And we subsequently built a convolutional neural network (CNN) model with the crystal graphs as input and the formation energy as output, which is used to achieve the prediction of the formation energy of the crystal material. Then we constructed the deep convolutional generative adversarial network (DCGAN) model on Bi-Se system and multicomponent system.
The next step is to integrate the prediction model into the DCGAN model as a back propagator following the constrained crystal DCGAN (CCDCGAN) scheme. After training, the built CCDCGAN model could automatically design thermodynamically stable structures. The generated structures are again relaxed by DFT calculation, and their formation energy are evaluated as well.
Results
We have got 10000 relaxed crystal structures of Bi-Se, Co-Hf systems as database, and we have used the trained CCDCGAN model to get one million Bi-Se structures, and more than 10000structures from various systems. And after the DFT calculation, we have found more than 100structures able to redefine the convex hull of Cd-Li, Mn-Se, Ni-Ta, Mn-Ta, Co-Li-O, Co-Hf systems. And CCDCGAN reproduced the experimentally known phases even if they are not in the training database.
Discussion
CCDCGAN designs crystalline materials that do not exist in the database and are thermodynamically stable. We further find that the performance of the model improves by increasing the number of crystal structures learnt, which demonstrates the potential of the model in the design of new structures. The model can therefore be used to search for new thermodynamically stable crystalline materials, thus speeding up the development of new crystalline materials.