First-Principles Calculations on Grain Boundaries in Nanocrystalline Graphene
 Perera, Delwin_First-principles calculations on grain boundaries in nanocrystalline graphene
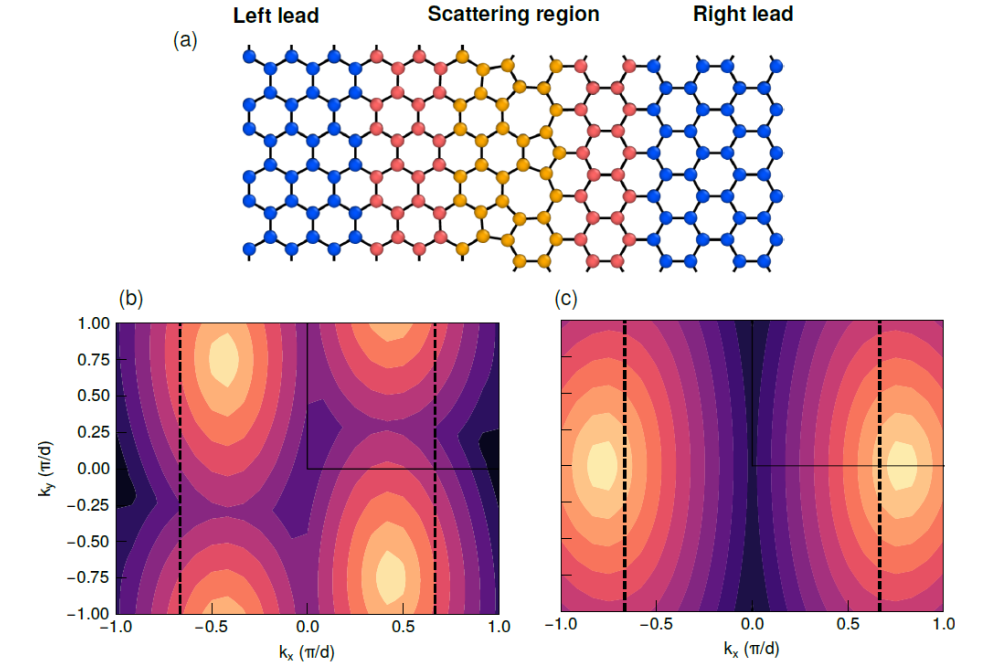
Perera, Delwin_First-principles calculations on grain boundaries in nanocrystalline grapheneFigure 1: Transport calculation setup and electronic structure of graphene bicrystals. (a) Setup for an armchair/zigzag bicrystal [(5, 0)|(3, 3)]. (b), (c) Contour plots of the top most valence band in the twodimensional Brillouin zone of two differently orientated graphene grains [(5, 3)|(7, 0)] under a tensile strain of 10% along the ky axis. The resulting supercell is only periodic along the kx direction. The bright spots indicate the positions of the Dirac cones and the dashed black lines show the position of the Dirac cones along the kx axis in the unstrained case.
Delwin PereraIntroduction
The modification of intrinsic properties of graphene via defects, such as grain boundaries, has been an intense field of research in the last years. The large scale production of graphene, for example by chemical vapor deposition, inevitably produces grain boundaries. Grain boundaries seem to be a particularly influential defect type in graphene as they affect the electronic properties quite notably. They have been found to induce transport gaps which turn graphene from a semi-metal into a semiconductor. Moreover, it has been shown that mechanical strain can both induce transport gaps and alter their magnitude. Along these findings it was recently demonstrated that nanocrystalline graphene (NCG) exhibits a more pronounced piezoresistive behaviour than single- or microcrystalline graphene. With this project we try to understand this finding based on electronic structure calculations. A thorough investigation of the relationship between grain boundary structure and electromechanical properties will aid to optimize the strain sensing capability of NCG.
Methods
Our simulations are primarily based on density functional theory (DFT) in conjunction with the non-equilibrium Green function formalism. Since these methods solve the electronic structure problem they have a high demand of computational resources and require high performance computers for systems with more than a few atoms. Tight binding calculations supplement our DFT calculations allowing to treat systems of much bigger size compared to DFT at the same computational cost.
Results
The first project period (2018) focused on the influence of the GB structure on electron transmission in graphene bicrystals with a fixed misorientation angle. We found that a transport gap arises for this misorientation angle, due to a momentum mismatch of the Dirac cones of adjacent grains. However, no dependence of the gap size for different structural realizations at the GB was observed. In the last project period (2019) we have generalized the idea of the momentum mismatch condition to arbitrary misorientation relations. Moreover, we have included the influence of mechanical strain which triggers a modulation of the gap size.
Discussion
The momentum mismatch condition for electron transport in graphene bicrystals originates from the location of the Dirac cones of individual grains since electron transport around the Fermi energy is most relevant. Electrons are only propagated in the overlapping region of the Dirac cones and any resonances outside this overlap region do not contribute to the transmission. The momentum mismatch condition implies a generalization for the calculation of the transport gap size and its modulation by mechanical strain solely based on the pristine graphene band structure. This has been studied in detail in the current project period resulting in a description of the transmission properties for arbitrary misorientation relations and two dimensional mechanicalstrains. The last part of the project will deal with a modified transport setup in which electrons are injected via a probe to a graphene nanocrystall with hexagonally shaped grains.