Modeling of Sortase-Linker Interactions for the iGEM Competition
Introduction
Sortase A7M is an enzyme, which catalyzes the linking reaction of the N-terminal and the Cterminal region of two proteins. To improve the efficacy of this reaction we set out to model the structure of the enzyme, understand the mechanism and improve the binding to the active site of by mutating the linkers used.
Methods
Our first step was to model the 3D structure of the enzyme using RosettaCM. RosettaCM is a comparative modeling algorithm implemented in the RosettaCommons software package. The next step was to identify the active site. To this end, we employed molecular dynamics simulations using the GROMACS software package. In the last step we once again used RosettaCommons to perform docking simulations to find the best possible linker.
Results
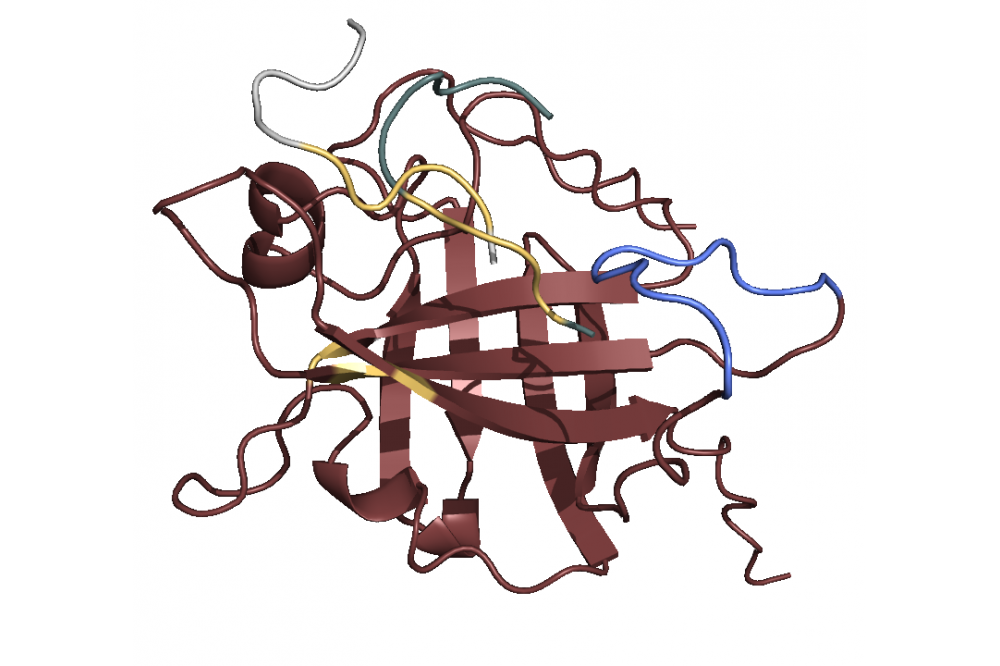
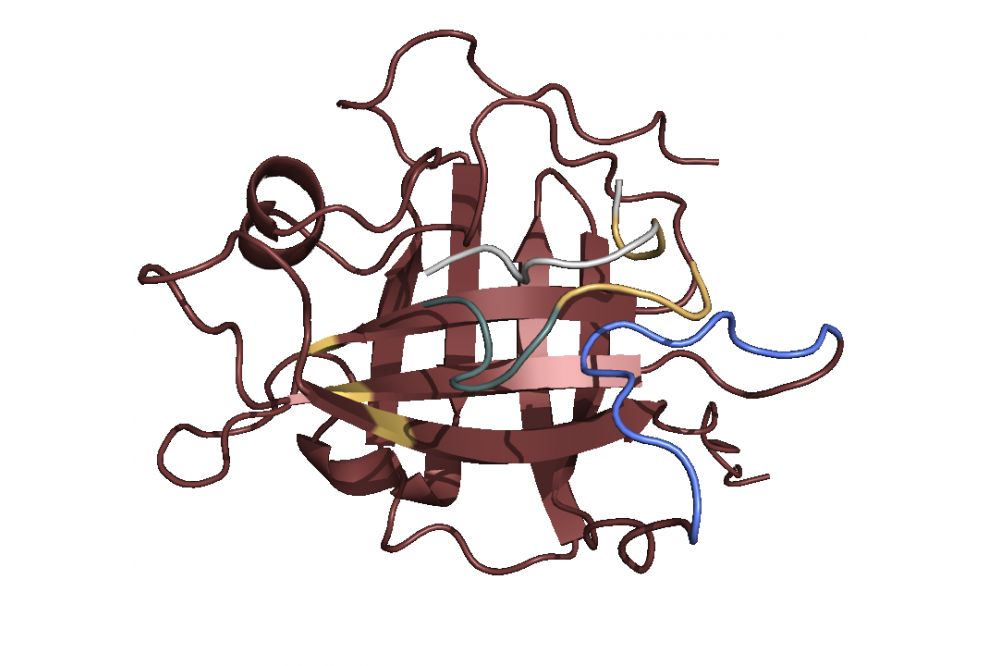
After modeling the structure we used principal component analysis to find the active site. The analysis revealed a Sortase substructure called β7/β8 loop to be crucial for the mechanism as literature confirms [1]. This substructure seems to move towards and away from the active site. We performed docking simulations and found out that the C-terminus with the specific LPETGmotif favors binding to the active site 1, while the N-terminus with a poly-G motif (GGGGG) favors binding to the loop 2. Combining this finding with the insights concerning the loop movement, we postulate that the loop transports bound ploy-G peptides towards the active site, where the LPETG peptides are located. This leads to the reaction and ultimately to the linkage. In terms of linker optimization we were not able find sequences with better binding affinities as most linkers achieved a similar binding score.
Discussion
With the computations performed we were able to model a Sortase A7M structure and postulate a mechanism. While we were not able to find ideal linkers for the reaction the insights we gained helped us develop a better understanding of the mechanism. To fully understand the mechanism and find linkers that are better suited for our reactions, we would need to perform hybrid molecular dynamic/quantum mechanics simulations as those would not only consider binding interactions but also reactions.