Quantum Chemical Investigation of Spectroscopic Properties of Iron Complexes as Models for Fe-N-C Fuel Cell Catalysts
Introduction
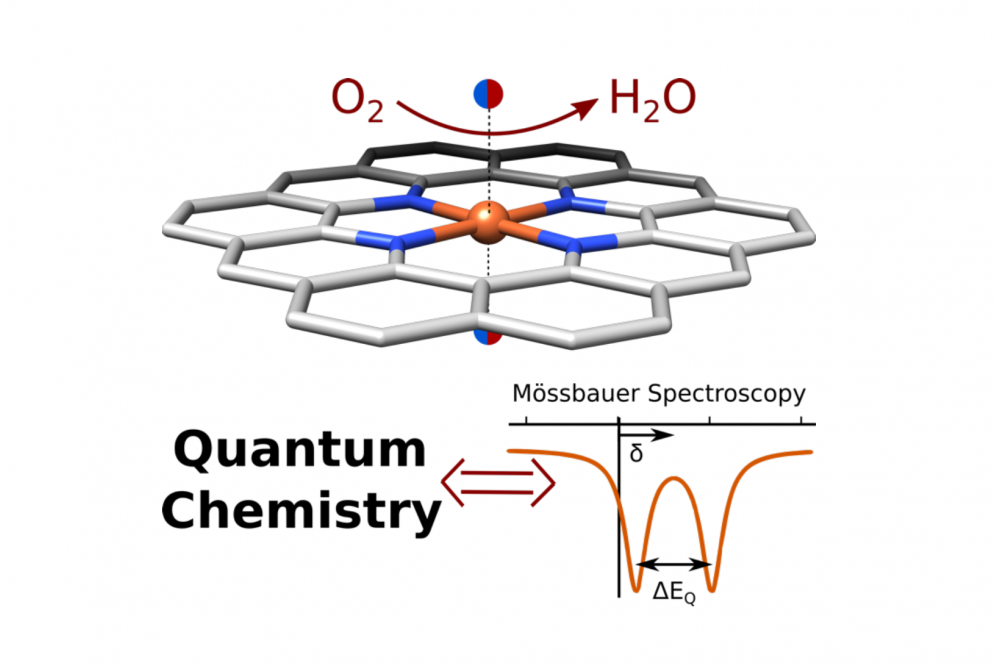
Fe-N-C catalysts are a promising substitution material for rare and expensive platinum catalysts on the cathodic side of proton exchange fuel cells. Their application could make the fuel cell technology widely accessible and contribute to a more climate-friendly automotive sector. However, Fe-N-C catalysts still lack stability and a fundamental understanding of their active site and the mechanism of oxygen reduction reaction has not yet been obtained. By combining spectroscopy and quantum chemical calculations this project aims to uncover the active site of Fe-N-C catalysts and to contribute information on its electronic structure.
Methods
This project mainly uses density functional theory calculations (DFT) to compute spectroscopic properties of molecular models aiming to mimick the active site of Fe-N-C catalysts and comparing them to experimental results provided by collaborators. During the screening process towards finding a suitable model, the structures are chosen in a way that different structural effects are investigated systematically and occurring phenomena can be explained. The main focus lies on the prediction of characteristic Mössbauer parameters, but also other spectroscopic techniques are targeted in a later stage of the project. Additionally, for a more precise description of the electronic structure, relevant structural models will be investigated by more elaborate (and computationally more demanding) methods such as CASSCF/CASPT2.
Results
An extensive calibration study on computational Mössbauer spectroscopy was performed in the first stage of the project. It was found that three different density functionals can predict relevant Mössbauer parameters, namely the isomer shift and the quadrupole splitting, with good accuracy. Furthermore, it was found during the 2D screening of catalyst candidates that the model size had a non-negligible impact on the electronic structure and relative spin state energies. Before proceeding to more complex model structures, e.g. 3D models, this effect needs further investigation.
Discussion
Future work will focus on expanding the screening of 2D and 3D model structures for Fe-N-C catalysts. Furthermore, the use of more accurate methods (CASSCF/CASPT2) is planned for the investigation of promising models from the DFT screening process.