Analysis of Magnetically Coupled Wavefunctions in Transition Metal Complexes
Introduction
Magnetic interactions in transition metal complexes arise from coupling of unpaired electrons, usually localized primarily on the metal ions. They dominate the electronic structure description but are difficult to predict solely by empirical rules derived from experimental data. Therefore, quantum mechanical calculations of the electronic structure are used to aid in the interpretation of experimental data and analyse the underlying principles of magnetic coupling. Solving electronic structures of such a high level of complexity requires the use of high performance computers.
Methods
Traditionally, computational chemistry methods based on density functional theory (DFT) have been used to describe magnetically coupled systems, first and foremost the so-called 'brokensymmetry' variant of DFT. Although it can be applied to a wide variety of chemical systems, this method is limited to certain representations of the wavefunction and cannot describe the individual rungs of the spin ladder that arises from magnetic coupling between spin centres. In recent years, density matrix renormalisation group (DMRG) has become available to the chemical community, which permits the description of complicated electronic structures and, most importantly in this context, the calculation of individual spin state wavefunctions.
Results
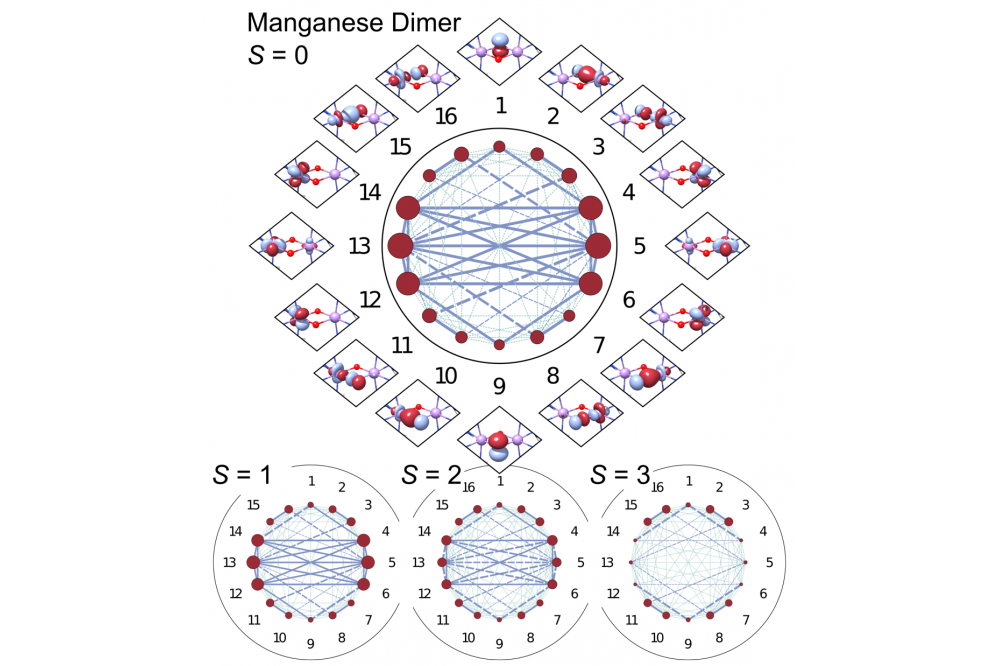
In this project, we developed a new approach for analysing magnetically coupled systems based on descriptors readily available in the DMRG method. We applied DMRG to several manganese dimers and analysed the resulting electronic structures based on the obtained orbital entanglement measures. We showed that these orbital entanglement measures, specifically the single-orbital entropy and the mutual information, offer a good representation of the magnetic coupling topology and are sensitive to chemical changes in homologous complexes.
Discussion
The approach developed in this project provides a better understanding of magnetically coupled systems than available through density functional theory. In the next project period, we plan to apply it to different types of chemical systems to explore its capabilities and limitations.