Global Optimization of Aluminum-doped Tin Clusters
 Rivic, Filip_Global Optimization of Aluminum-doped Tin Clusters Project ID
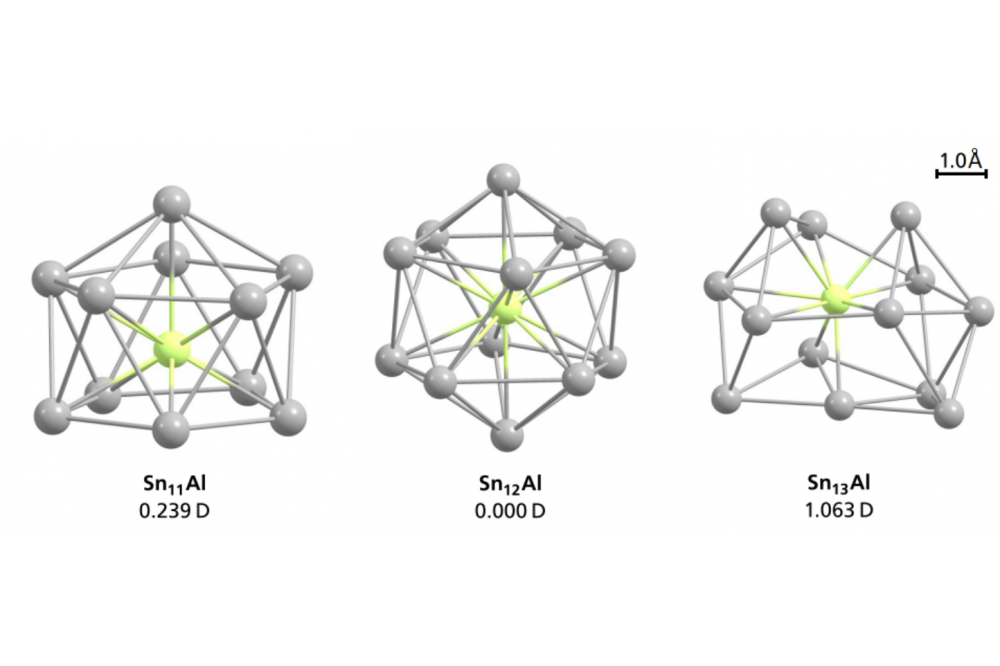
Rivic, Filip_Global Optimization of Aluminum-doped Tin Clusters Project IDFigure 1: Predidcted state structures and the corresponding calculated dipole moments for SnNAl with N=11 – 13 at the PBE0/def2-TZVPP level of theory. Tin atoms are displayed as gray spheres while the aluminum atom is shown in green.
Filip RivicIntroduction
Semiconductors are the fundament of modern technology and as such still a topic of current research. With decrease in size, opto-electric properties change significantly and the investigation of materials at the sub-nanoscale become mandatory. By changing the size and chemical composition of nano-clusters band gaps can be tuned and typical metal like tin becomes semiconductive. Tin clusters are known to form particularly stable cages which are suitable hosts for any kind of doping. Aluminum-doped tin clusters are the microscopic counterpart to positive-doped bulk semiconductors. Quantum chemical calculations are crucial to understand the formation process and the geometrical structure of these nanosystems. Furthermore, the calculations allow insight into the electronic structure and dielectric behavior. Predicted geometrical structures can be further compared with experimental data generated by electric and magnetic molecular beam deflection experiments.
Methods
The theoretical prediction of the geometrical structure is carried out with the help of a genetic algorithm on plane-wave density functional theory level. Calculations for all clusters are performed with an applied Ultrasoft-Rabe-Rappe-Kaxiras-Joannopoulos-pseudopotential for both elements and an energy cut-off of 30 Ry. Electrons which are explicitly described are calculated with a Methfessel-Paxton-smearing. Convergence is assumed if the energy of the global minimum is constant within 1000 new calculated structures. These calculations are performed using the Quantum Espresso software package. In the next step structural candidates in an energy range of 2.00 eV relative to the global minimum are further optimized. These optimizations are carried out with NWChem on spin unrestricted Gaussian-orbital based DFT (PBE0/def2-TZVPP). The programs ORCA and JANPA were used for the calculation of vibrational frequencies and population analyses employing the same level of theory.
Results
Within this project we globally optimized the geometrical structures of SnNAl with N = 8 – 16. Furthermore, we calculated vibrational frequencies, dipole moments and performed population analyses for isomers within an energy range of about 0.1 eV relative to the energy of the predicted global minimum. These candidates are compared to experimental data. It was possible for most of the investigated clusters to successfully assign the results generated by molecular beam deflection experiments to the calculated geometry of the structural isomer with the lowest energy. However, for SnNAl clusters with N = 12 a much higher dipole moment was observed in the electric beam deflection experiment compared to the theoretical predictions.
Discussion
The final globally optimized structures for SnNAl with N = 8 – 16 in this project suggest a similar growth pattern to transition metal doped tin clusters. Electric beam deflection experiments confirm this for most species. In case of the Sn12Al-cluster the calculated geometries suggest a slightly distorted icosahedron still possessing a center of inversion. With this non-polar structure, a vanishing dipole moment was predicted. Therefore, no beam broadening should be observed. However, the experimental data suggest a polar structure with a dipole moment. Even energetic higher lying isomers and structures with initially guessed symmetries were investigated and compared to the experimental data but no match could be found. In the further process, the binding energies were calculated and compared to the other SnNAl clusters. Here, the Sn12Al-cluster shows an exceptionally high binding energy and stiffer vibrational modes than any other aluminum-doped tin cluster. In future projects we want to extend the quantum chemical study of these clusters with calculations of density of states and band gaps. Additionally, we want to employ other methods like DDEC6 and Bader charge partition analysis to get a closer insight into the electronic structure of the Sn12Al-cluster. Simultaneously, we want to investigate similar p-doped tin systems like gallium-doped tin clusters which could be interesting due to similarities in the electronic structure.