Redox Chemistry of Ternary Graphite Intercalation Compounds
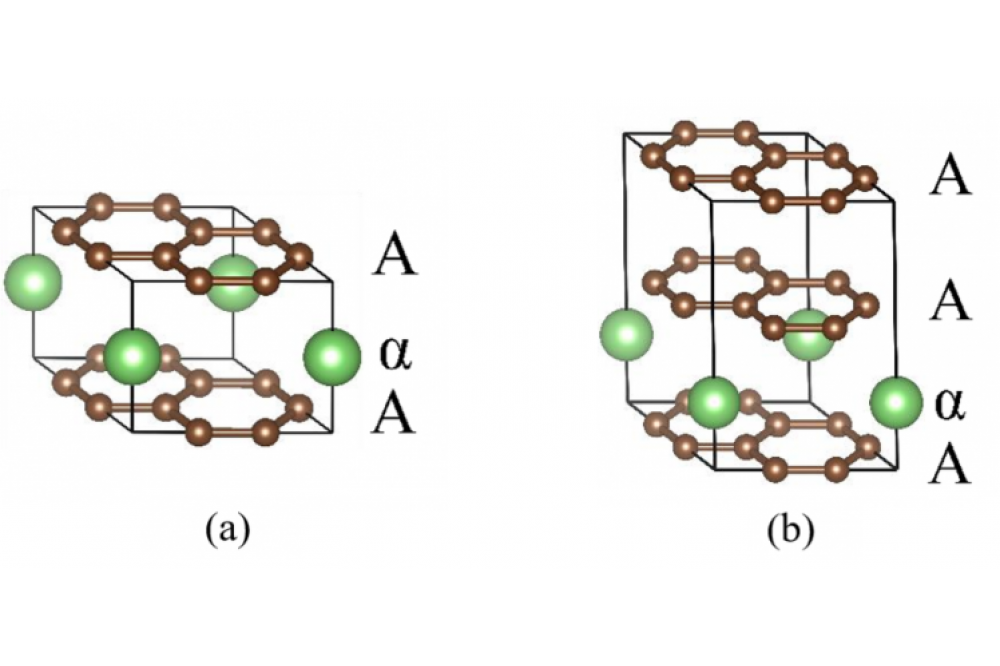 Lenchuk, Olena, Redox chemistry of ternary graphite intercalation compounds, Figure 1
Lenchuk, Olena, Redox chemistry of ternary graphite intercalation compounds, Figure 1Figure 1: Atomic structure of the (a) stage-I LiC6 and (b) stage-II LiC12 compounds.
Olena Lenchuk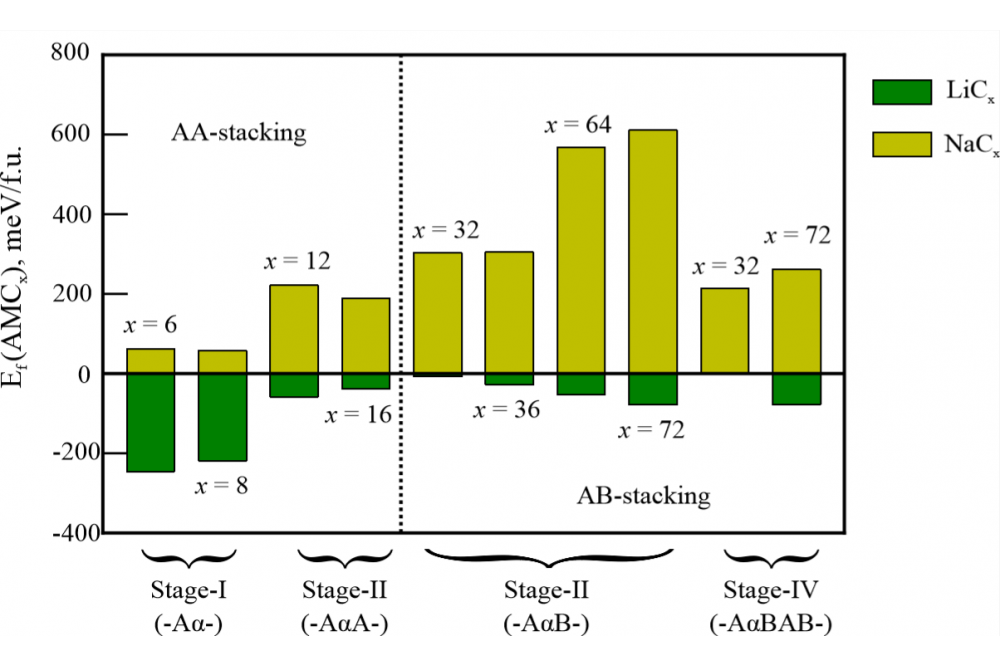 Lenchuk, Olena, Redox chemistry of ternary graphite intercalation compounds, Figure 2
Lenchuk, Olena, Redox chemistry of ternary graphite intercalation compounds, Figure 2Figure 2: The calculated formation energies of AM-GICs (AM = Li, Na) for different alkali metal concentrations (6 ≤ x ≤ 72) and the lowest-energy graphite layer stacking. The black dashed line separates the AM-GICs compounds with the AA- (left-hand side) and AB- (right-hand side) stacking preference of the graphite layers.
Olena Lenchuk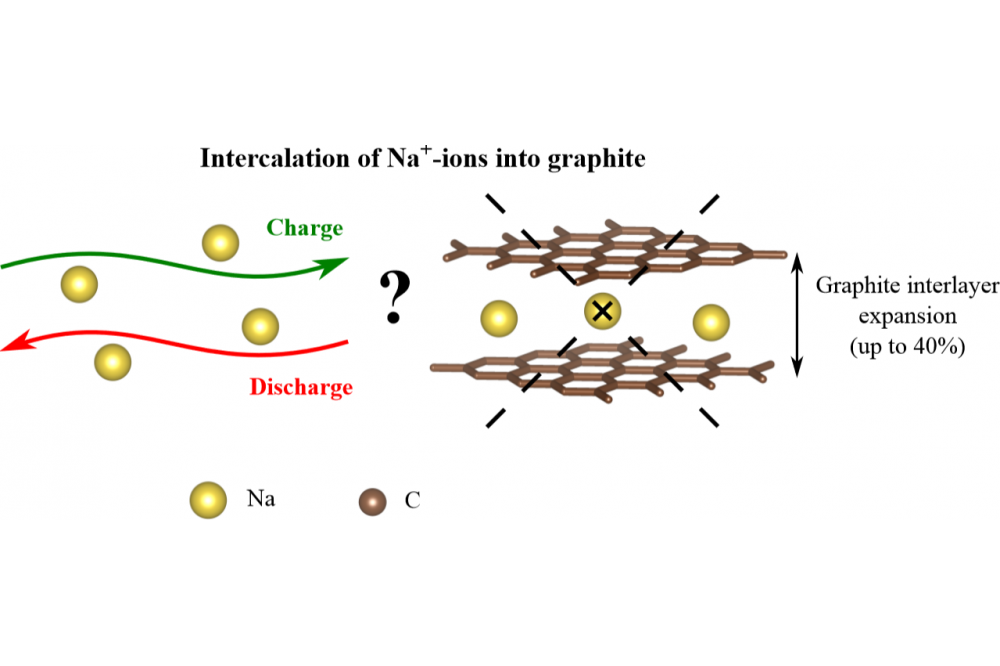
Introduction
Sodium-ion batteries (SIBs) are considered as alternatives to lithium-ion batteries (LIBs), showing a potential in reducing the material cost and advancing from the same charge storage mechanisms as in LIBs [1-5]. The current project is aimed to investigate the structure and thermodynamic stability of alkali metal - graphite intercalation compounds (AM-GICs), which are formed during alkali metal intercalation into graphite. The thermodynamically stable GICs with high AM-content possessing the composition of AMC6 or AMC8 exist for lithium (LiC6), potassium (KC8), rubidium (RbC8) and cesium (CsC8) [6]. For sodium, however, only few low-concentration Na-GICs have been reported [7-8]. Na-GICs compounds are still a matter of debate. The working hypotheses of the observed behaviour are related to the structural deformation of the graphite during sodium intercalation as well as to the differences in the binding nature between alkali metals and graphite. Here, we present a small part of our study.
Methods
In our research, we first focused on the systematic investigation of the structure and energetics of the binary GICs at different stages (from stage-I to stage-IV) and various concentrations of the intercalated lithium and sodium ions (from AMC6 to AMC72). For this purpose, we performed density functional theory (DFT) [9-10] calculations with periodic boundary conditions. For the investigated models, dispersion correction to the DFT energy (DFT-D3 method, proposed by Grimme et al. [11-12]), was used for the description of the van der Waals interactions in AM-GICs. The alkali metals were located on top positions (denoted as “α” in Figure 1), whereas the AB- and AA-stacking of the graphite interlayers was considered. Atomic structures of the investigated AMC6 and AMC12 compounds are shown in Figure 1.
Results
The thermodynamic stability of different AM-GICs was evaluated by means of the formation energy, shown in Figure 2. Our results suggest that a transition from AB- to AA-stacked graphite takes place upon increasing the alkali metal concentration. This transition is observed for stage-II AM-GICs in the concentration range between x = 16 and x = 32. This finding is also consistent for the investigated alkali metals, lithium and sodium.
Discussion
Negative values of the formation energy (see Figure 2) indicate that binary GICs are stable with respect to the AB-stacked graphite and the bulk alkali metal. Our results indicate that the intercalation of Li into graphite is always energetically preferred. The driving force for Li intercalation depends on the mutual distribution of Li atoms (its concentration and stage). The formation energy of Na-GICs also varies depending on the concentration of Na atoms, but in contrast to Li, intercalation of Na atoms into graphite is always determined to be thermodynamically unfavorable. The analysis of the bond distances in Li- and Na-GICs shows that stretching of the in-plane C-C bonds in the investigated compounds is negligibly small compared to the pristine graphite and the observed volume expansion upon AM intercalation is caused by the expansion of the graphite interlayers. Furthermore, the graphite interlayer distances are increased upon increasing the atomic radius of the intercalated alkali metals, namely up to 15 % for lithium and up to 40 % for sodium. The observed results are in reasonable agreement with experimental data [7-8] and other theoretical studies [13-14].
Outlook
In this report, we have presented calculations of the structure and thermodynamic stability of Li- and Na-graphite intercalation compounds by means of DFT calculations. Our results reveal that, in contrast to Li-GICs, Na-GICs are always thermodynamically unstable. The energy contributions considering the structural deformation and the binding strength in the compounds affect the stability of the Na-GICs [15].