Large-Scale Biophysics with Hartree-Fock
 Luc Wieners_Large-Scale Biophysics with Hartree-Fock_Figure1
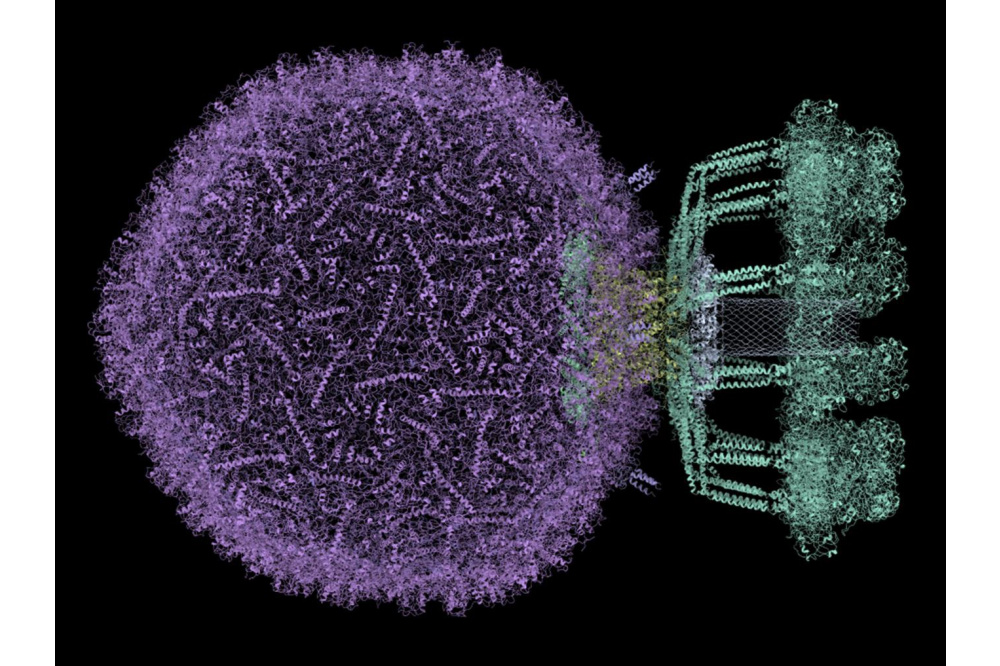
Luc Wieners_Large-Scale Biophysics with Hartree-Fock_Figure1Figure 1: The bacteriophage P68 (PDB 6Q3G) which was studied at quantum-mechanical accuracy in this project.
Luc Wieners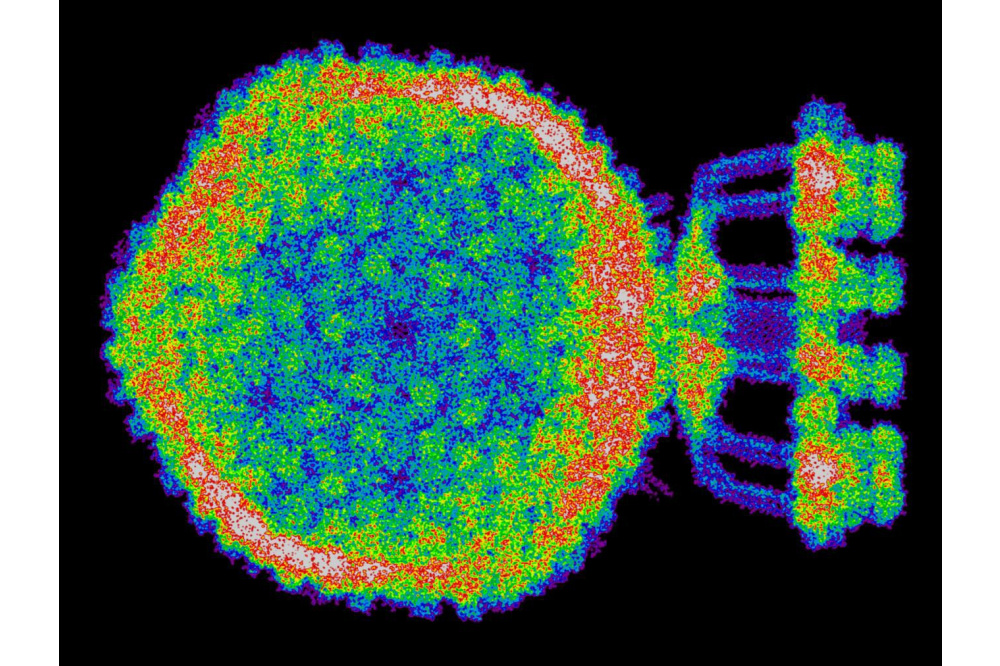 Luc Wieners_Large-Scale Biophysics with Hartree-Fock_Figure2
Luc Wieners_Large-Scale Biophysics with Hartree-Fock_Figure2Figure 2: Projection of the computed electronic density of the bacteriophage P68.
Luc WienersIntroduction
Quantum-mechanical calculations are needed for highly accurate investigations of molecules. They allow a subatomic treatment of matter by explicitly describing the electrons in the system. Due to the high computational complexity of these calculations, high performance computing is necessary to study larger systems. Biological structures, such as proteins, are still often considered out of reach for quantum-mechanical calculations. We therefore present a new algorithm that enables a quantum-mechanical treatment of systems from biophysics.
Methods
Our formalism uses the quantum-mechanical Hartree-Fock method. By combining improved algorithms with a divide-and-conquer approach, we developed a program to perform fast quantum-mechanical calculations which enable the investigation of proteins, protein complexes and DNA.
Results
As a result, we tested the formalism by calculating the electronic structure of multiple systems from structural biology, including a bacteriophage (Staphylococcus aureusphage P68, PDB 6Q3G), a virus that attacks bacteria, in a solution of water. This system contains over 600 individual proteins and more than 45 million atoms and is, to our knowledge, the largest system to be investigated at quantum-mechanical accuracy using the Hartree-Fock method. To perform the calculation, the structure was divided into over 240,000 individual clusters for which an independent calculation was performed. The calculations were then brought together to create a three-dimensional map of the electronic structure. The bacteriophage and its computed electron density can be seen in the corresponding pictures. We also performed calculations of predicted protein structures by AlphaFold, a program that recently made an immense breakthrough in the field of protein folding for which the developers were awarded with the Nobel prize in chemistry 2024. Despite providing highly accurate predictions for protein structures, AlphaFold still sometimes has uncertainties and therefore assesses the validity of its own predictions by providing a so- called confidence score. We compared quantum-mechanical calculations to assessments from AlphaFold and found a strong correlation between atomic energies and structure confidence scores. This leads to a new metric for evaluating predicted protein structures based on methods from first principles which could help to gain new insights in the prediction of protein structures. Finally, we added the option to calculate spectral properties of molecules to our program which enables the calculation of absorption spectra of biomolecules previously out of reach, containing a few hundred to a few thousand atoms. As example systems we studied the anti-cancer drug Actinomycin D, DNA at different lengths and Actinomycin D bound to DNA. The calculated spectra show good agreement with experimental results and therefore are an additional validation of the developed formalism.
Discussion
The presented project opens new opportunities for calculations in structural biology, quantum biology, theoretical chemistry, and material science, especially for studying systems previously out of reach. Additionally, follow-up projects could investigate proteins under electric and magnetic fields or in excited states, providing more insights into the dynamics of proteins