Learning by Building: Construction of Voltage-Dependent Ion Channels from Modular Components II
 Jan Krumbach_Learning by Building: Construction of voltage-Dependent Ion Channels from Modular Components II_Figure1
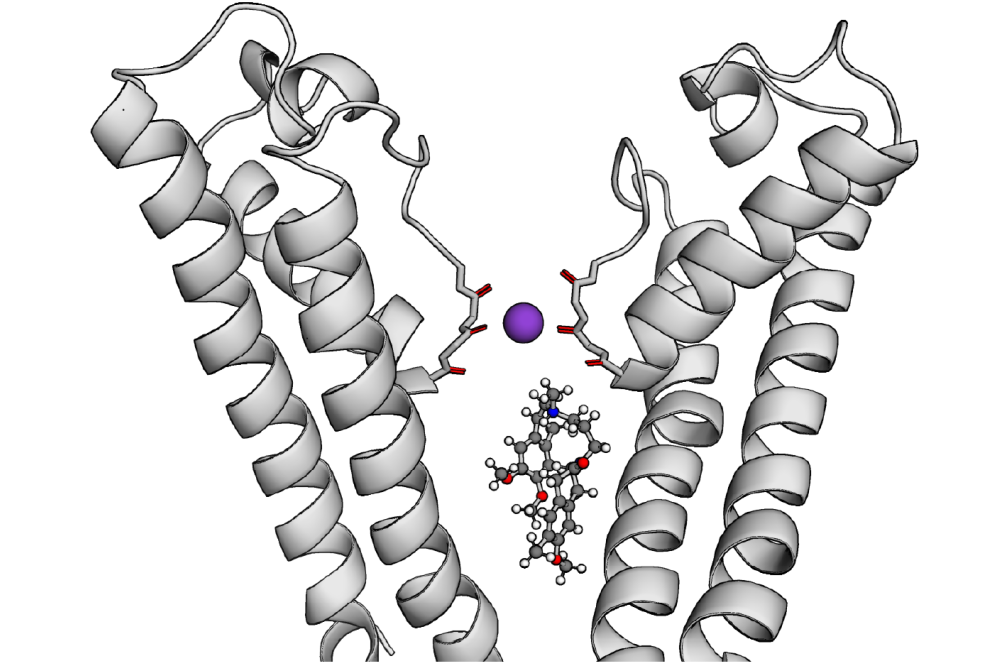
Jan Krumbach_Learning by Building: Construction of voltage-Dependent Ion Channels from Modular Components II_Figure1Figure 1: Depiction of HCN4 pore region with the channel inhibitor ivabradine below the K+-occupied selectivity filter (SF). The protein is depicted as gray cartoon, with carbonyl oxgens within the selectivity filter shown in the stick repre- sentation. A violet sphere represents the K+ cation in the lower SF, while ivabradine is depicted in the pore below in the ball-and-stick representation (with the nitrogen atom in the functionally relevant tertiary ammonium group colored in blue). Only two opposing monomers of the tetrameric channel are shown for visual clarity.
Jan KrumbachIntroduction
Voltage-dependent K+ channels are facilitators of a diverse set of physiological processes, including neuronal signaling and viral infection cycles. Consequently, potassium channels are highly relevant therapeutic targets for drugs. Fundamental research regarding structural-functional relationships will likely help to aid these efforts. On the most basic level, canonical K+ channels consist of a central pore, which contains the selectivity filter (SF) that allows the selective conduction of potassium. Conformational changes in regulatory domains, sensitive to voltage or ligands, close and open the central pore in response to external stimuli. Two potassium channels are studied as part of this project: The synthetic KvSynth1 and HCN channels. KvSynth1 is a chimeric channel, consisting of the small, constitutively conducting viral K+ channel Kcv PBCV-1 as pore domain and the Ciona intestinalis phosphatase voltage- sensing domain. The resultant chimeric protein is a functional K+ channel and additionally voltage-gated, thus combining characteristics of both building blocks. In contrast, HCN are naturally occurring complex K+ channels, that play a central role in human and animal physiology, most notably due to their involvement in the generation and modulation of the pacemaking current in heart cells. HCN channels exhibit a dual gating pattern, with a predominant voltage-gate, but additional modulation of the open state by binding of cAMP as ligand. With ivabradine, an inhibitor of HCN channels is already commercially available as remedy for the heart condition Angina pectoris. The transmission of mechanical signals during gating is studied in this project for both channels with coarse-grained models. Additionally, the molecular mechanism of HCN channel blockade by ivabradine is elucidated.
Methods
3D structures are a necessary pre-requisite for any protein dynamics computation. The structure of KvSynth1 is predicted using both homology modeling approaches as well as neuronal network-based structure prediction methods. For HCN channels, resolved Cryo-EM structures, including a newly resolved HCN4 structure with the channel blocker ivabradine within the pore, are used as basis for simulations. A multi-scale approach to simulate protein dynamics is employed, consisting of both MD simulations and elastic network models (ENM). In MD simulations, Newtonian equations of motion are solved numerically with a detailed set of parameters for inter-atomic interactions to simulate dynamics in atomistic resolution. In contrast, single amino acid residues are modeled as vertices in the coarse-grained ENM model, with covalent and non-covalent interactions between spatially adjacent residues modeled as harmonic springs. MD simulations allow the study of dynamics in high temporal and spatial resolution, while large-scale, slow conformational changes of protein domains during voltage-gating can be studied with ENM based methods. Both methods are used complementary to elucidate channel dynamics: Global rearrangements of KvSynth1 during voltage-gating and of HCN channels upon ligand-binding are modeled with ENM-based methods, whereas the quality detailed dynamics of the inhibitor ivabradine in the HCN pore are retraced with MD simulations. Neural network-based structure prediction and MD simulations require considerable computation time and can benefit greatly from GPU-acceleration. For these crucial steps of our work, HPC resources provided by the Lichtenberg high performance cluster are essential.
Results
For HCN1 and HCN4, allosteric coupling between ligand-binding domains was observerd in computations with ENM-based methods. These predicted allosteric interactions are in line with more recent experimental findings. Starting from the first available resolved cryoEM structure with the small molecule inhibitor ivabradine in the pore of HCN4, the key role of the charged tertiary ammonium for channel blockade by retaining K+ cations in the lower SF above was elucidated. A charge in this position was necessary to maintain the blockade in simulations, which strongly indicates electrostatic repulsion with the K+ cation in the SF as main mechanism of blockade. This observed charge dependence of the blockade on a molecular scale is also concordant with the experimentally verified current-dependence of the HCN blockade by ivabradine. Hydrohobic amino acid residues, that can interact with the aromatic rings of ivabradine to anchor the positively charged functional group below the SF, were also identified.
Discussion
Fundalmental research of mechanical transmissions between regulatory and sensory sites in potassium channels could inform the development of remedies against ion channel related diseases, so called channelopathies. For ivabradine, knowledge of detailed mechanisms of interaction within the HCN channel pore could inform additional research seeking compounds with increased specificity for HCN4 in the heart over other HCN channels to mitigate side effects. In subsequent project periods, additional structures of KvSynth1 will be generated by both homology modeling as well as NN-based methods and combined approaches for an increased model fidelity. Protein dynamics of these generated structures will be analyzed by the set of computational methods already successfully applied to HCN.