On the Protonation of Verdazyl Species in Aqueous Acidic Electrolytes
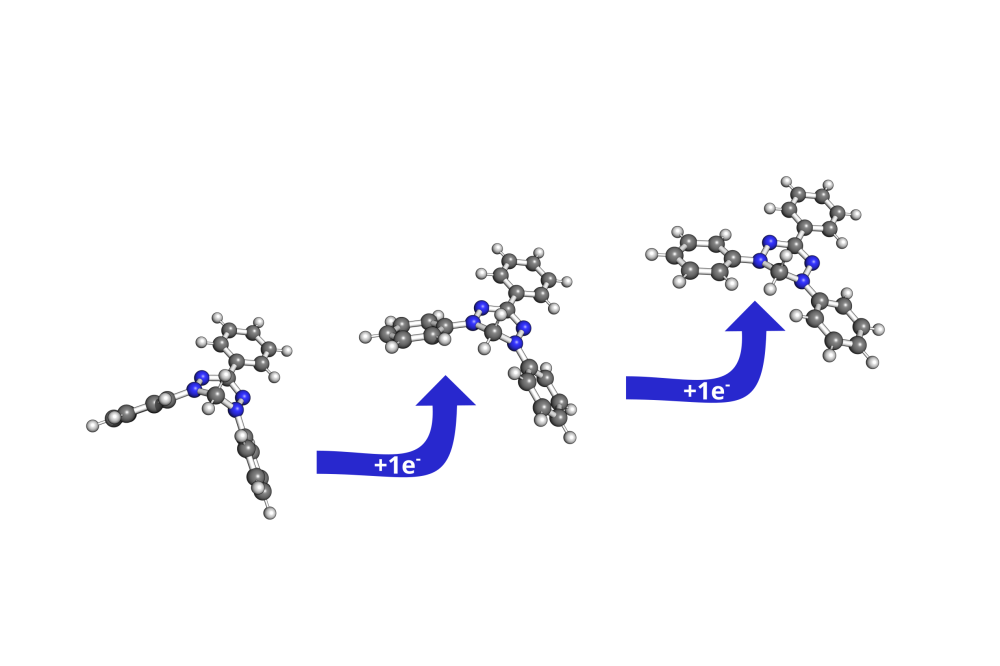 Pietruschka_Dennis_S._OntheProtonationofVerdazylSpeciesinAqueousAcidicElectrolytes_Figure1
Pietruschka_Dennis_S._OntheProtonationofVerdazylSpeciesinAqueousAcidicElectrolytes_Figure1Figure 1: The structural deformation of the 1,3,5-triphenyl verdazyl cation a) by addition of one electron b) and two electrons c) is shown. The structures were optimized at the B3LYP-D4/def2-TZVPD level of theory, including solvation effects via COSMO (εr = 78.3553).
Dennis S. PietruschkaIntroduction
Verdazyl radicals are discussed as a possible candidate for application in organic redox flow batteries. These species are characterised by their stability and sterically unprotected radical moiety as well as a desirable redox-behavior.[1] In an interdisciplinary approach, combining electrochemical characterization, synthesis and quantum chemical computations, we characterised the triphenly verdazyl species in an aqueous, acidic environment after disproportionation.[2] A two-electron-step was identified to take place in a strongly acidic environment (pH < 3). The computational studies supported the experimental approach by providing a rationale for the observation: In a sufficiently acidic medium a doubly protonated verdazyl species was identified as energetically favourable. In accordance, this species is also suggested by mass spectrometry. The ansatz of combining tight-binding methods (xTB) and density-functional theory allowed for the successful identification of the most favourable protonation sites of the molecule. Strong structural deformations were observed as a consequence of the introduction of two protons to the anionic verdazyl species.
Methods
We initially performed an automated protonation site screening[3] for the identified product of the disproportionation reaction using the CREST program developed by the Grimme group.[4] We carried out an structural optimisation using the extended tight binding method xTB.[5] Four chemically distinct protonation sites were identified and considered in subsequent density functional theory (DFT) calculations. The calculations were carried out using the Turbomole programm package (version 7.5.2).[6] The systems were considered at the B3LYP-D4/def2-TZVPD level of theory employing the Conductor-like Solvent Model (COSMO) to account for the solvation effects of the aqueous medium.[7] The structures optimized by DFT were confirmed as local minima by frequency calculations. The thermodynamic corrections were used to calculate the Gibbs energies of reaction ΔrG that were used for the comparison of protonation reactions on the different sites of the molecule.
Results
A comparison of the Gibbs energies of reaction in solution revealed the unsubstituted nitrogen atoms of the verdazyl molecule to be the energetically most favourable protonation positions. Relative to the substituted tertiary nitrogen atoms other protonation sites, such as the carbons of the phenylsubstituents, are approximately 90 kJ/mol higher in energy. Overall the protonation is strongly exothermic, regardless of the surrounding medium’s pH-value. Acidic pH-values between 0 and 3 were considered.
A different picture, however, emerges for the second protonation step. Here, the last unsubstituted nitrogen atom is the most favourable protonation site. This time, the protonation reaction is only slightly exergonic in a strongly acidic medium. A turning point in the exergonicity is calculated at a pH-value of 2.6, where the reaction becomes endergonic.
Both protonation steps induce strong deformation in the molecular structure due to rehybridization effects, which was found to influence the absorption properties of the compound in solution.
These results were complemented by electro- and physico-chemical characterisation, presenting further evidence for the in situ formation of this previously unobserved species.
Discussion
An automated protonation site screening coupled with a DFT-based approach was able to present additional evidence for the formation of doubly protonated verdazyl species, obtained from disproportionation in a strongly acidic medium. We were able to present a thermodynamic picture of this protonation mechanism. An upper limit of the pH-value where this two-step protonation takes place was identified and is in agreement with experimental observations.
Furthermore, the structural characterisation of the protonated and doubly protonated verdazyl species revealed strong structural deformations as a consequence of rehybridization induced by the introduction of additional protons to the central verdazyl ring structure. These electronic and structural effects present a rationale for the observed changes in the absorption spectra during cell-operation.
The presented two-electron mechanism coupled with fast redox kinetics motivates further research of this promising material class of substituted verdazyl radicals.