Intermolecular London Dispersion Interactions of Azobenzene Switches
 Intermolecular London Dispersion Interactions of Azobenzene Switches
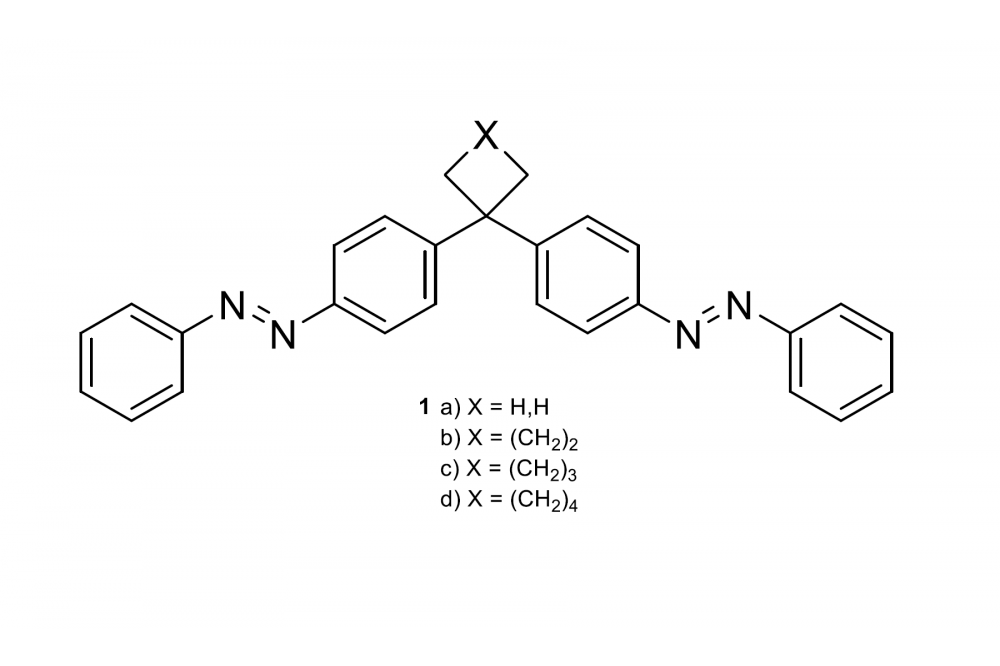
Intermolecular London Dispersion Interactions of Azobenzene SwitchesFig. 1: Bis‐azobenzenes 1 with different connectors.
Anne KunzIntroduction
Azobenzenes (AB) are promising candidates for molecular solar thermal energy storage (MOST) systems. After synthesizing different bisazobenzenes 1 a-d with different alkyl-substitution on their bridgehead, differential scanning calorimetry (DSC) measurements were performed. It revealed, that intermolecular attractive London dispersion interactions stabilize the (E)-isomer in bisazobenzene, due to the different alkyl bridges leading to a higher storage enthalpy. This computational study was performed to support the obtained experimental data. The focus was laid on the storage enthalpies between the all-(Z)- and (E)-isomers.
Methods
For this project Gaussian16 was used to conduct our computations. All structures were optimized at the PBE0 density functional theory level with D3(BJ) dispersion correction using the def2‐TZVP basis set. Molecular structures were visualized using the CYLview software.
Results
By combining the experimental and computational data it was suggested, that in the all-(E)-isomer of compounds 1 a-d there is an intermolecular attractive interaction of the azobenzene with the cycloalkane bridge due to London dispersion. Even though the AB moieties in all molecules investigated are electronically equal increasing London dispersionstrength by different bridging units increased the energy difference between (E)- and (Z)-state. Those interactions will serve as a basis to design future MOST systems.
Outlook
Future investigations will also be conducted with support from accompanying computational studies on the Skylla Cluster Gießen.