Computational Modelling of Multiple Photoswitches
Introduction
The computational modeling of molecular transformations in organic chemistry, e.g. photo-inducible isomerizations of azobenzenes allows one to predict and design novel photoswitches and their reaction pathways. Molecular switches are of high importance for biology and material science, and computational modelling of their properties is an important step prior to the synthesis and experimental investigation.
Methods
Molecular geometries and thermochemical, as well as spectral properties were computed using Density Functional Theory (DFT) and time dependent DFT such as the PBE0 or BHLYP functionals. Highly precise energies were obtained using ab-initio DLPNO-CCSD(T) with at least triple zeta basis sets.
Results
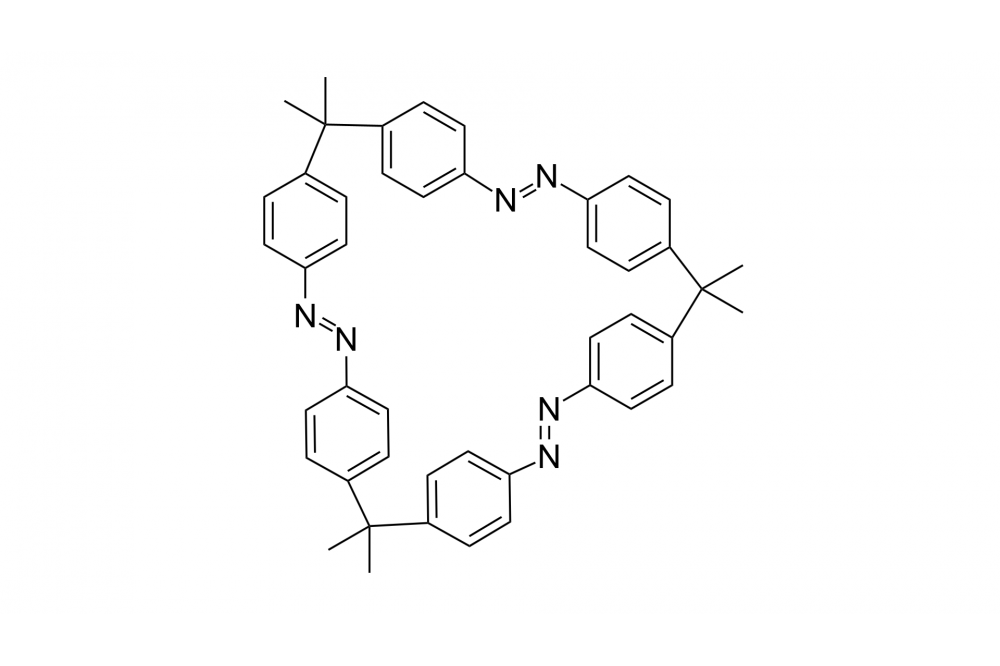
Several macrocycles containing multiple azobenzene units were synthesized and investigated. It was found that macrocyclic ring strain strongly influences the compounds thermal Z→E isomerization behavior, that could be used to for three-state photoswitching in a symmetric molecule. Furthermore, computations revealed that overcoming ring strain determines the rate determining step, making this unconventional switching behavior possible.