Metal-Free Dimerization of Terminal Alkynes Catalyzed by a Pyridonate Borane Complex
 Metal-free dimerization of terminal alkynes catalyzed by a pyridonate borane complex
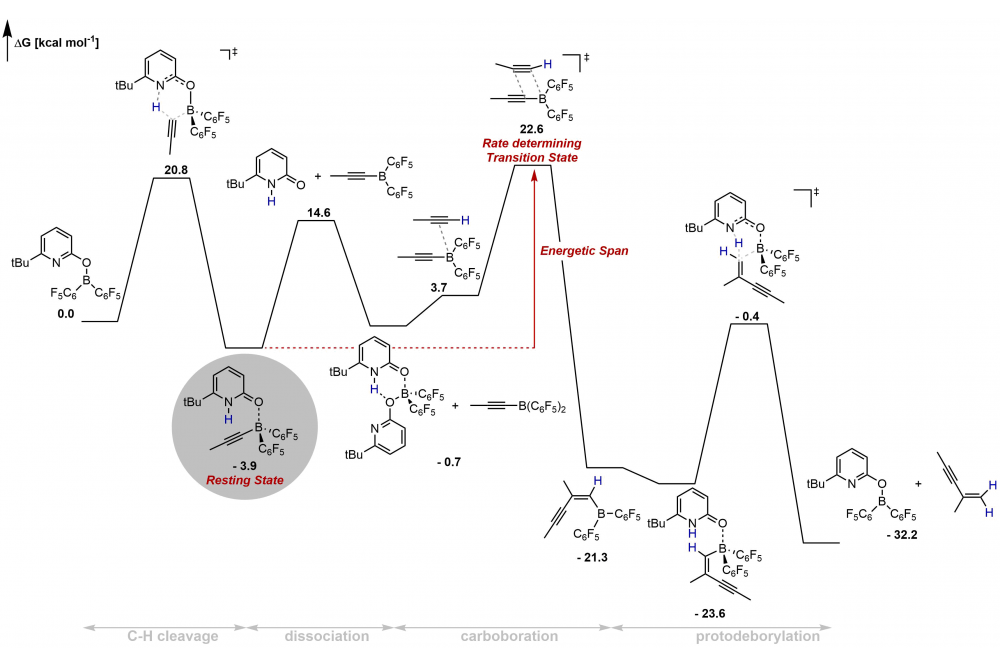
Metal-free dimerization of terminal alkynes catalyzed by a pyridonate borane complexFigure 1: Potential free energy surface of the gem selective dimerization of terminal alkynes catalyzed by a pyridonate borane complex.
Urs GellrichIntroduction
We have recently reported the synthesis of a pyridonate borane complex that reversibly activates dihydrogen under mild conditions. Significantly, computations and experiments show that the boroxypyridine undergoes a chemical transformation to a neutral pyridone donor ligand during the hydrogen activation. This unprecedented mode of action enabled us to develop the first metal-free gem selective dimerization of terminal alkynes by combining experimental investigations with computational chemistry.
Methods
The structure of all stationary points that are involved in this catalytic transformation were optimized at the density functional theory level (DFT) with the PBE0-D3(BJ)/def2-TZVP method. Solvent effects were taken implicitly into account using the SMD model for benzene. Thermodynamic properties were obtained at the same level of theory from a frequency calculation. Single-Point energies were computed with an accurate correlated ab inito method at the DLPNO-CCSD(T) level. All ab inito computations were done with the triple zeta def2-TZVP basis set. TightPNO settings were used. The computed free energies were corrected with regard to the standard state by adding RT ln(c0s/c0g) (i.e., about 1.84 kcal/mol) to energies of all structures. The NMR computations were done with the PBE0 functional and the pcS-2 basis set by Jensen, specifically designed for the computation of shielding constants, using the GIAO method.
Results
The mechanism of the metal-free gem selective dimerization of terminal alkynes catalyzed by a pyridonate borane complex was investigated experimentally and with accurate TightPNO-DLPNO-CCSD(T)/def2-TZVP//PBE0-D3(BJ)/def2-TZVP computations. The catalytic transformation commences with C−H cleavage by a boroxypyridne that displays frustrated Lewis pair (FLP) reactivity. Significantly, the boroxypridine undergoes a chemical transformation to a neutral pyridone ligand. The pyridone borane complex that forms upon C-H cleavage dissociates to a pyridone and an alkynylborane. The computations revealed a rare 1,2-carboboration of an alkyne effected by the alkynylborane as C−C bond-forming step. While we were not able to isolate the alkynylborane, a comparison of the experimentally obtained NMR spectra with the spectral signatures simulated by DFT allowed its unambiguous identification in solution. The pyridone alkynylborane complex that forms upon the initial Csp-H complex was computationally identified as the resting state of the catalytic transformation, which agrees with experimental observations.
Discussion
An interplay of computational chemistry and experimental investigations allowed us to disclose the unusual catalytic reactivity of a pyridonate borane complex and to develop a novel metal-free catalytic protocol, namely the gem selective dimerization of terminal alkynes.
Outlook
We are currently transferring the insights that we gained in this study to new projects that aim for novel metal-free synthetic protocols using pyridonate borane complexes as catalysts.