High Throughput Screening and Tuning of Magnetic Two Dimensional Materials
 Dr. Xinru Li, HighThroughputScreeningandTuningofMagneticTwoDimensionalMaterials
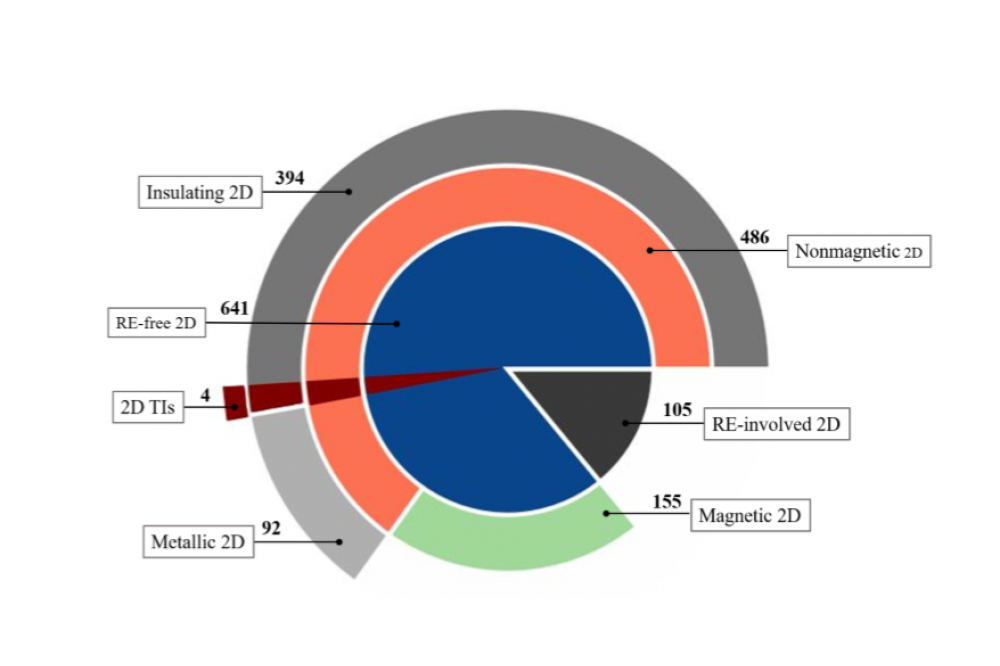
Dr. Xinru Li, HighThroughputScreeningandTuningofMagneticTwoDimensionalMaterialsFigure 1: Classification of 746 systems in the 2D materials database based on their electronic properties.
 Dr. Xinru Li, HighThroughputScreeningandTuningofMagneticTwoDimensionalMaterials1
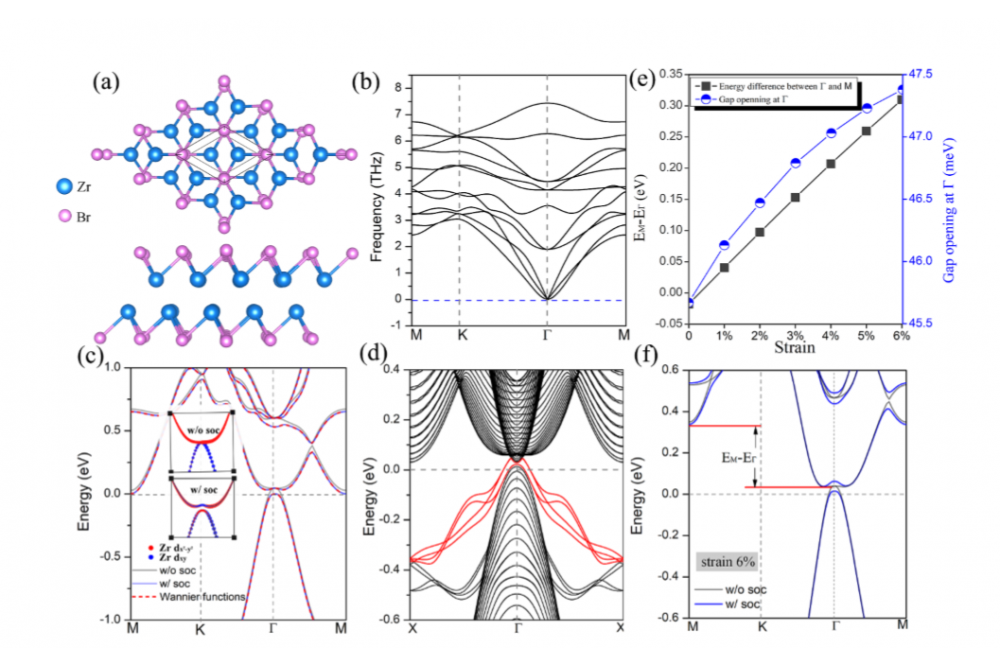
Dr. Xinru Li, HighThroughputScreeningandTuningofMagneticTwoDimensionalMaterials1Figure 2: (a) Top and side view of the crystal structures of ZrBr monolayers. (b) Phonon dispersions. (c) Band structure without SOC, with SOC and obtained from MLWFs indicated by gray solid line, blue solid line and red dashed line, respectively. Insets show the zoom-in view of the orbital resolved band structure near the Γ point. (d)Band structure of the1Dribbon with nontrivial edge states highlighted in red.
Xinru LiIntroduction
The demand of minimization and feasibility of integration makes two-dimensional (2D) materials remarkable in promising technological applications, whereas due to the breaking of translational symmetry, the 2D materials Display distinct properties than the corresponding bulk materials thus they are fundamentally interesting. Early in 2011, the US federal government has launched the Materials Genome Initiative, aiming at developing and exploring materials design strategically based on theoretical calculations using the well developed density functional theory methods, to go beyond the conventional way of materials discovery based on try-and-error or empirical knowledge. Regarding designing 2D materials, Ashton et.al. have searched for layered materials based on bulk crystal structures database Materials Project and predicted over 600 thermally stable 2D monolayers. However, such 2D materials have only been investigated by standard density functional theory calculations, without characterizing detailed functional properties. Hence, a thorough high throughput first principles study on 2D materials supported by high performance computational resources are of high demand. In our current project, for those nonmagnetic 2D materials in 2D materials database, the main guideline to the characterization is based on the accurate calculation of the band gap, resulting in the classification of 2D materials into possible photocatalysts, thermoelectrics or topologicalmaterialsandhencepredictionsforfutureenergyapplications. wehavesuccessfully classified These 2D materials according to electronic and magnetic properties by high throughput methods based on density functional theory.
Methods
The self consistent band structure calculations are carried out by using the augmented plane wave method as implemented in the VASP code. Spin polarized calculations with large initial magnetic moments are done in order to select the materials with nonmagnetic ground state. Moreover, the effect of SOC is considered self-consistently in all the calculations. The maximally localized Wannier functions (MLWFs) are constructed automatically with out implementation of a poor-man’s algorithm by using the Wannier90 code. The projectors are chosen by the integrated partial density of states (DOS), where only those orbitals with large enough weight around the Fermi energy are chosen. The energy window is determined by the number of projectors and total DOS, We define a descriptor Σ, which is essentially the least square difference function between the eigenvalues calculated by VASP and Wannier 90 at all k-points in the whole Brillouinzone,to Judge thequality of the MLWFs. It is observed Σ should be smaller than 0.01 in order to obtain reliable MLWFs. Afte rsuccessful constructions of the MLWFs,the Hamiltonian in the Wannier basis isused to evaluate the surface states and topologica linvariant.
Results
we have identified four topologically nontrivial two dimensional (2D) materials out of 641most stable systems in the 2D materials database. The HTP Screening is carried out based on the automated construction of maximally localized Wannier functions followed by explicit calculations of the surface states, where the Wannier centers are evaluated as a further verification of the nontriviality. The electronic structure of such non trivial materials is discussed in Detail together with the features in the crystal structure and relevant orbitals. The work about HTP screening of 2D TIs has been published in 2D Materials.