Global Optimization of Gadolinium Doped Tin Clusters
 Global Optimization of Europium-doped Tin Clusters
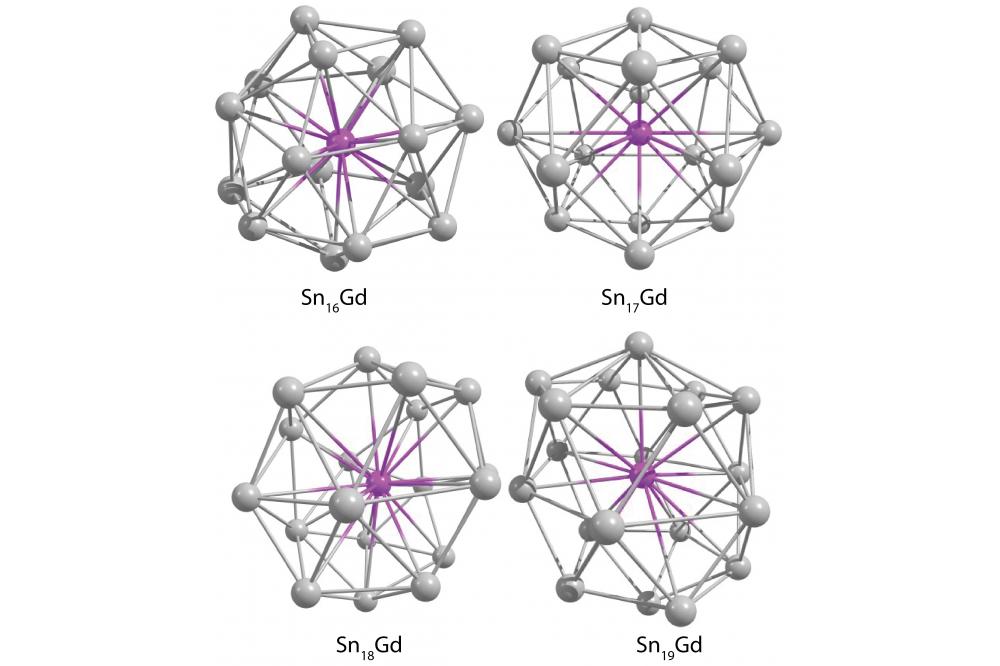
Global Optimization of Europium-doped Tin ClustersFigure 1: DFT ground state structures for SnNGd (N = 16 – 19) on the PBE0/def2-TVZPP level of theory.
Martin GleditzschIntroduction
Gadolinium-doped tin clusters belong to an interesting class of nanoalloys. They combine the quasi-aromatically stabilized structures of endohedral tin clusters with the versatile chemical properties of rare earth metals. The dopant gadolinium itself is unique due to its high amount of unpaired f electrons. These have an interesting effect on the tin cages, which amongst other things results in a different tin cage formation around the dopant as well as high electric dipole moments and electronic spin multiplicities of the overall cage structure. Such agglomerates might macroscopically assembled lead to a new class of materials, which could exhibit a diverse range of tunable properties. The discovery of fullerenes lead to the search of further cluster structures, which could macroscopically assembled behave different from there solid state counterparts. Here tin clusters are of interest, as their similar bonding pattern and the predicted spherical aromaticity suggest these to be possible inorganic analogues to fullerenes.
Methods
The experimental research on these sub-nano systems is challenging, as it is difficult to synthesize these small structures shielded from external influences. From theoretical perspective it is difficult to properly calculate the experimentally relevant structures, as this requires computationally expensive and thorough global optimizations. A chemists educated guess is biased, as structures in that range have unpredictable geometries and properties. In the past, we have proven the high quality of our molecular beam deflection experiments in several studies. The synergistic effect of combined experimental and theoretical investigations is well known in our group.
Results
The global minimum search via the genetic algorithm has been successfully performed. The resulting structural candidates for SnNGd with N = 6-8;16-22 have been further optimized with gaussian-orbital based density functional theory (DFT). Structural and dielectric properties have been calculated. Figure 1 exemplarly shows the predicted ground states for N =16-19 on the PBE0/def2-TZVPP level of theory.
Discussion
While doping of N = 6 - 9 tin atoms leads to the formation of exohedral complexes, with N= 16 - 22 this results in endohedral structures which fully encase the dopant. This behavior is expected, as only large tin clusters have been previously shown to encapsulate metal dopants. The smaller species with N = 6 -9 seem to mimic the neutral or anionic tin clusters with the dopant being capped further away. This results in relatively high absolutes of the electric dipole moment. The encapsulation of the gadolinium for N = 16 - 22 leads to highly symmetric structures with far lower absolutes of the dipole moment. While the ground states of N = 16, 17 closely resemble manganese-doped tin clusters, N=18-22 represent new structures. The resulting theoretical data will be compared with electric and magnetic beam deflection experiments to assign the experimental structures.