High-Fidelity Calculations of Charge Transition Levels and Formation Energies of Point Defects in Strontium and Barium Titanate
 Anton Volodin_High-Fidelity Calculations of Charge Transition Levels and Formation Energies of Point Defects in Strontium and Barium Titanate_Figure1
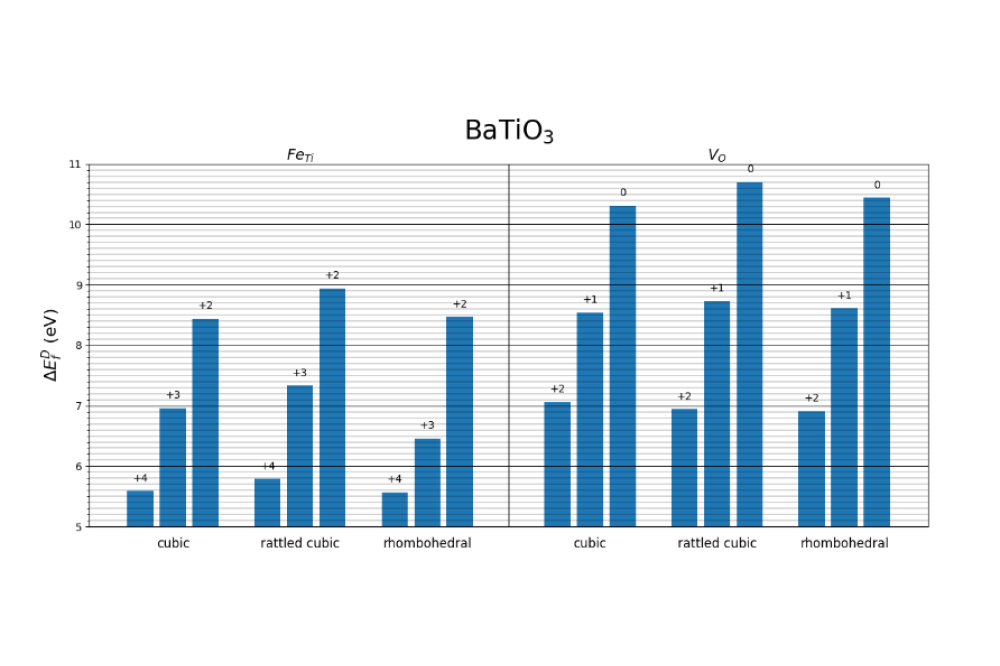
Anton Volodin_High-Fidelity Calculations of Charge Transition Levels and Formation Energies of Point Defects in Strontium and Barium Titanate_Figure1Figure 1: Formation energies of defects in different charge states computed using cubic, rattled and rhombohedral phases of BaTiO3 as reference. Left: formation energies of Fe substituting Ti defect, iron has oxidation states 4+, 3+, 2+. Right: formation energies of oxygen vacancy with charge states 2+, 1+, 0.
Introduction
Electroceramic materials are integral to modern technology, with applications ranging from capacitors and temperature sensors to solid electrolytes and varistors. In most cases, ceramic materials are doped to achieve the desired performance. However, doping introduces multiple possible compensation mechanisms, and researchers often rely on a trial and-error approach when developing electroceramic materials with doping. This reflects a broader lack of understanding of the fundamental principles governing defect physics in electroceramics. To address this gap, ab initio calculations primarily using density functional theory (DFT) - are increasingly employed to investigate defect behavior. Barium titanate (BTO) and strontium titanate (STO) are two widely studied electroceramic materials, best known for their role as dielectric materials in capacitors. Both are model perovskite systems and have been extensively examined for their unique soft-mode physics and phase transitions. Nevertheless, DFT modeling of these materials poses challenges due to the dynamic instability of their cubic phases at 0 K - the temperature at which DFT calculations are performed. Dynamic instability means that on the potential energy surface the system is located on top of the hill, where the derivative of energy with respect to displacement is zero. This means that the forces are zero, and ions are not moving during structure relaxation. However, introduced defect acts as a disturbance, pushing the system off the hill on potential energy surface and inducing extensive ionic relaxations all over the simulation cell. In the end, a cubic perovskite system is compared with a distorted system, leading to errors in computed defect formation energies and discrepancies between theoretical results and experiment. The aim of this project was to evaluate strategies for accounting for the dynamic instability of BTO and STO. The investigated defects were oxygen vacancy and iron substitution on titania site, as these are common defects in these materials.
Methods
The formation energies of defects were computed within the density functional theory (DFT) formalism using Vienna ab-initio Simulation Package (VASP). Using rattled structures as reference states for computing defect formation energies, and applying symmetry constraints during DFT calculations are the two approaches to account for dynamic instability of BTO and STO considered. Rattled structures are created by slightly displacing ions in random directions. After relaxing the rattled structure, it is not located on top of the hill anymore, but is being brought to a local minima. Symmetry constraints prevent the massive ionic displacements from taking place when a defect is introduced in the system. During structure relaxation, only those motions of ions that conserve the symmetry group of a system are allowed.
Results
Using rattled structures instead of conventional perfect cubic perovskite structures leads to an increase in total energies computed by up to 1.1 eV. Defects formation energies are increased by up to 3.7 meV/atom.
Discussion
Overall, the impact of the applied strategies on the values of defect formation energies computed is only mild, not increasing several hundreds meV. However, it is still noticeable. The 500 meV increase in formation energies leads to change in concentration of defects of approximately 6⋅10-5 at 600 K. The impact of applying symmetry constraints is less pronounced, as it shifts defect formation energies by only up to 200 meV. Hence, the effect of rattling and constraining symmetry is significant and should be considered when computing defect formation energies with DFT. Further investigation is needed to further evaluate the effectiveness of the strategies.