Defect chemistry and Band structure of NBT-xBT
 Hu_Pengcheng_Defect chemistry and Band structure of NBT-xBT_Fig1
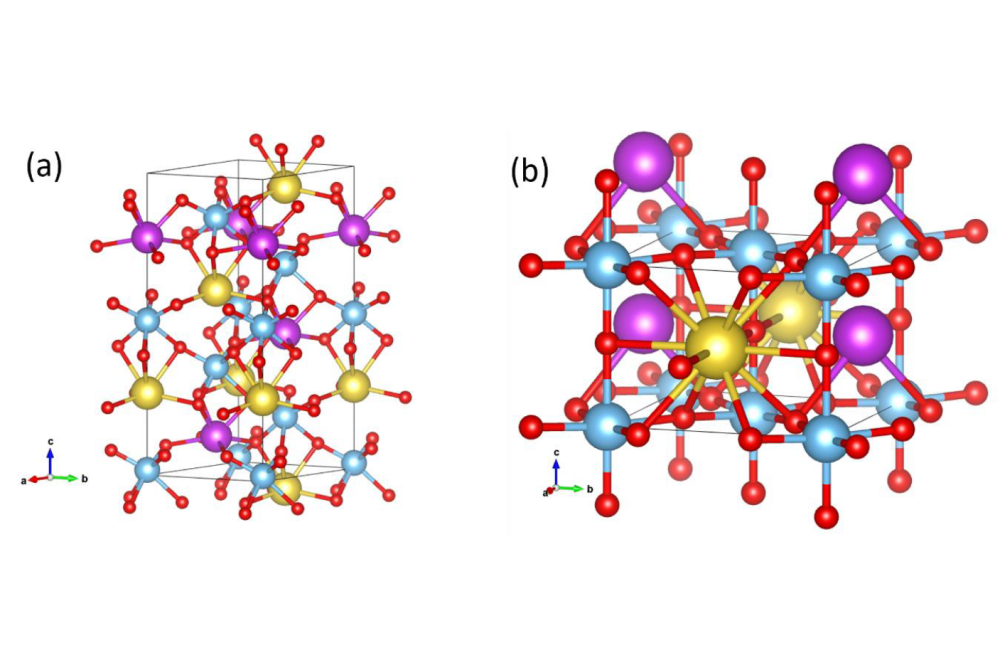
Hu_Pengcheng_Defect chemistry and Band structure of NBT-xBT_Fig1Figure 1. (a) The rhombohedral and (b) tetragonal NBT used for the defect formation energy calculation.
Pengcheng Hu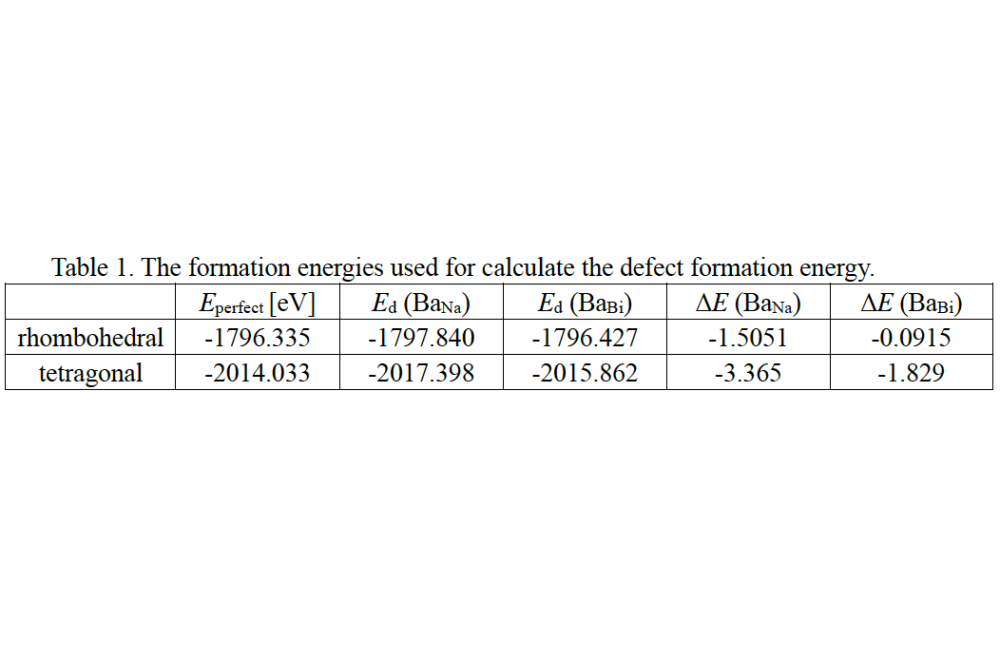 Hu_Pengcheng_Defect chemistry and Band structure of NBT-xBT_Table1
Hu_Pengcheng_Defect chemistry and Band structure of NBT-xBT_Table1Table 1. The formation energies used for calculate the defect formation energy.
Pengcheng HuIntroduction
Sodium bismuth titanate-barium titanate (Na0.5Bi0.5TiO3-BaTiO3, abbreviated as NBT- BT) with high Curie temperature and a remnant polarization is considered to be an important material for the fabrication of lead-free piezoelectric materials. However, one major drawback of NBT-xBT as a piezoelectric/dielectric material is its high electrical conductivity which leads to high and unacceptable dielectric loss and leakage currents at elevated temperatures. Improved understanding of electrical conductivity and the conduction mechanism(s) of NBT is essential to solve this problem.
According to our experimental data: we are measuring electrical properties of NBT-BT with different BT content. It seems that for BT contents up to 6%, NBT-BT seems to be an n-type electronic conductor, while we get p-type electronic conduction for NBT-9BT. It seems like the Ba prefers to substitute for Na for low BT content, i.e. in orthorhombic NBT, and prefers Bi substitution in tetragonal NBT. It can be easily solved by the calculation of the defect formation energy of BaNa and BaBi in (maybe only with 111-cation ordered) rhombohedral and tetragonal NBT.
Band structure: according to our experimental data: The electrochemical reduction (electron trapping) in NBT-BT piezoceramics was studied by in-situ XPS measurements. And the metallic Bi starts to develop when the binding energy of the Bi 4f7/2 reaches about 159.7 eV.
It is necessary to determine the CBM of NBT-xBT. We assume that the CBM of NBT is mainly derived from Bi 6p and Ti 3d states, while BT has mainly Ti 3d character. Therefore, both the Ti4+ and Bi3+ could be reduced, which determined by the relative position of charge transition level of Ti4+/3+ and Bi3+/0. But it needs calculation to prove it. Such calculations of defect chemistry and band structure need to be performed using density functional theory (DFT) calculation and can help to understand the instability of
electrode interface and electrical conductivity and the conduction mechanism(s), which could improve the performance of NBT-based multilayer actuators, transducers, and ceramic capacitors. The density functional theory (DFT) calculation has to be done with high-performance computing (HPC) systems.
Methods
For all total energy calculations, we use the Vienna Ab initio Simulation Package (VASP). We applied the projector augmented waves method, using the generalized-gradient approximation (GGA) in the Perdew-Burke-Ernzerhof parameterization. For the valence electron configuration, we chose O: 2s22p4, Na: 2p63s1, Ti: 3s23p64s23d2, Bi: 5d106s26p3, and Ba: 5s25p66s2. The k-point mesh and the cut-off energy obtained from Vaspkit software Most results have been obtained with the open-source code Abinit. In this case, we used norm-conserving PBEsol pseudopotentials from the website PseudoDojo. Except for Na, where we additionally integrate the 2p states, we use the same valence electron configuration as given above. All structures have been visualized with the open-source software Vesta. We use this equation to calculate the formation energies of defects (BaNa and BaBi) and employ different structural phases of NBT to simulate the NBT-xBT system. Specifically, rhombohedral NBT is used to represent compositions with lower Ba content (x < 0.06), while tetragonal NBT is used for higher Ba content (x > 0.06). The defect formation energies are then calculated for each case to investigate the relationship between Ba content and its preferred substitution site.
E = Edefect − Eperfect − μBa + μNa/Bi
Results
The results indicate that Ba atoms preferentially substitute at the Na site in both rhombohedral and tetragonal NBT. The calculated formation energies of BaNa and BaBi are -0.092 eV and -1.505 eV, respectively, in rhombohedral NBT, and -1.829 eV and -3.365 eV, respectively, in tetragonal NBT. However, these findings appear to conflict with the electrical conductivity measurements: for BT contents up to 6%, NBT-BT exhibits n-type electronic conduction, whereas at 9% BT (NBT-9BT), the material shows p-type conduction.
Discussion
Calculating defect formation energies in NBT using density functional theory (DFT) presents challenges due to the intrinsic A-site disorder caused by the random distribution of Na and Bi atoms in the actual material. In contrast, DFT simulations require a predefined A-site ordering, which has been shown to substantially affect the computed defect formation energies. Therefore, the observed electronic conductivity behavior cannot be attributed to Ba substitution. Instead, we propose that it may result from an effective background donor effect. In the case of 9% BT content (NBT-9BT), this could be due to the substitution of BaṄa and antisite defect of BiN̈a and oxygen vacancies. For lower BT contents (less than 6%), it could be the substitution of BaBi and antisite defect of NaB̈i and A-site or B-site vacancies.