Multiscale Modelling of Cement-Based Materials III
 Izadifar_Mohammadreza_Multiscale Modelling of Cement-Based Materials III_Fig1
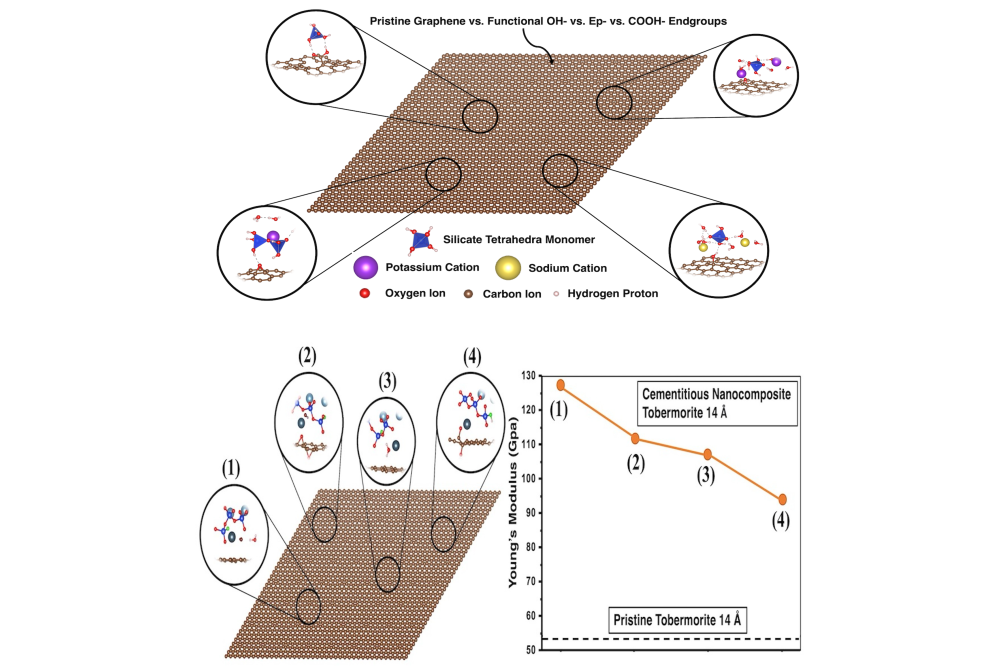
Izadifar_Mohammadreza_Multiscale Modelling of Cement-Based Materials III_Fig1Figure 1: (A) Geopolymer nanocomposites incorporating graphene-based nanomaterials in the presence of (OH-), epoxy (Ep-), and carboxyl (COOH-) groups as a contribution of defects on its surface. (B) Cementitious nanocomposite of tobermorite 14Å with intercalated hydroxyl/rGO and epoxy/rGO layers.
Mohammadreza IzadifarEinleitung
The project seeks to create a comprehensive model linking nanoscale phenomena to larger-scale behaviors to better understand the chemical reactions in cement-based and geopolymer materials with graphene/based nanomaterials. This effort involves employing atomistic simulations such as ab-initio (DFT) and molecular dynamics (including meta-MD and ReaxFF) to investigate fundamental processes. Larger-scale modeling techniques, like atomistic kinetic Monte Carlo (kMC) and Coarse-Grained Monte Carlo, are used to assess and confirm reaction rates and surface chemistry.
Methoden
These projects utilize atomistic modeling techniques through the Vienna ab initio simulation package (VASP) for equilibrium density functional theory (DFT) implemented for various configurations. Molecular Dynamics (MD) simulations with ReaxFF are conducted using the LAMMPS package. Moreover, CGMC simulations were carried out using the canonical ensemble (NVT), which maintains a constant number of particles, volume, and temperature.
Ergebnisse
In [1], a fundamental understanding of the interactions between graphene-based nanosheets and geopolymer performed, focusing on the monomer units, namely, Al(OH)4-.K+, SiO(OH)3-.K+.3H2O, SiO2(OH)22-.2K+.6H2O, as well as dimerized structures of Si2O(OH)6, and Si2O2(OH)5-K+. 3H2O, using equilibrium DFT with and without the contribution of vdW dispersion interaction, along with ReaxFF-MD computational modeling approaches. In [2], we proposed four structural models with the same stoichiometry of calcium, silicon, and carbon atoms. We studied this kind of rGO composite with and without interstitial water molecules to investigate the expected changes in their elastic properties. First, we performed calculations without the contribution of interstitial water molecules in order to compute the elastic constants of the cementitious nanocomposite with tobermorite 14Å within the DFT computational approach. Moreover, the elastic constants of four more structural models identical but now with the contribution of 7 interstitial water molecules have been investigated. In [3], we implemented a 3D off-lattice CGMC simulation of a coarse-grained model for studying the nucleation of alkaline aluminosilicate gel for three different silicate- activated systems. Based on the Gibbs free energy of dimerization for four different monomer species of four different monomer species of Si(OH)4, Al(OH)4−.Na+, SiO(OH)3−.Na+.3H2O, and SiO2(OH)22−.2Na+.6H2O, and considering the limitation of the tetrahedral structure for the particle polymerization in the system, an 3D off-lattice CGMG approach is adopted. Each monomer species is represented as a distinct coarse-grained particle type to compute the gel structure evolution as a function of the different number of iterations. In this manner, three different pH values (11, 12, and 13) have been investigated, incorporating a larger simulation system to obtain more accurate results using Octree cells expansion. In [4], we investigated the dynamical adsorption of a nanodroplet of water on the graphene-coated geopolymer surface. It will deeply focus on the analysis of the interfacial adhesion between a graphene layer and metakaolin geopolymer surface as well as the hydrophobicity and the strength of the modified geopolymer surface. Molecular dynamics (MD) and DFT calculations are carried out to evaluate the surface and the adsorption energies for low and high silicon content. The surface wettability characteristics is examined from the contact angle between a nanodroplet of water on the graphene-coated surface. The water diffusion is further analyzed for the non-coated and coated geopolymer nanostructure.
Diskussion
It has been observed that The interaction of Si(OH)4 aqueous specie with the pristine graphene substrate (i.e. without defects) presents small physical adsorption due to the dipole nature of this overall neutrally charged specie. When the quantity of sodium or potassium cation, involved into the system, increases, the adsorption energy of Si monomer unit on the pristine graphene substrate increases substantially. The adsorption energy of Al(OH)4 in the presence of sodium is more favorable in comparison with the contribution of potassium cation for the adsorption on all the substrate cases (G, G-OH, G-O). This agrees with the higher surface charge of Na+, resulting in stronger electrostatic interaction [1].
Upon relaxing the model structures, we observed that the dissociation of hydroxyl functional groups from the hydroxyl/ rGO lattice took place not only in the presence of Ca2+ ions for charge compensation but also when the Ca2+ charges were compensated with hydroxyl groups. On the contrary, rGO/CSH interactions remained consistent with the initial structural models of the epoxy/rGO surface. It has been observed that the elastic constants for all the models were positive, so that the proposed structures are indeed stable [2].
Systems with pH levels of 11 and 12 exhibit notably similar behavior regarding the quantity of monomers at the simulation's conclusion, displaying a marginal difference of merely 0.67%. Conversely, the system with pH 13 showcases a higher monomer content by the simulation's conclusion, depicting a 32.87% increase compared to the pH 11 system and a similar 32.20% increase in contrast to the pH 12 system [3].
According to our observations, it is evident that the mobility of water molecules on the coated surface was twenty times lower, with a diffusion coefficient of 0.131x10-10 m2.s-1. Importantly, our study revealed the crucial role of interfacial chemical bonding, with the evaluated elastic and indentation moduli increasing 70 and 39 times, respectively, when the interfacial bonding concentration was 14.15%. Our findings provided fundamental insights into the interactions between graphene-coated geopolymer surfaces and water nanodroplets, representing a crucial step towards the development of superhydrophobic geopolymer materials [4].