Principles of On-Surface Synthesis
 Simone Sanna_Principles of On-Surface Synthesis_Figure1
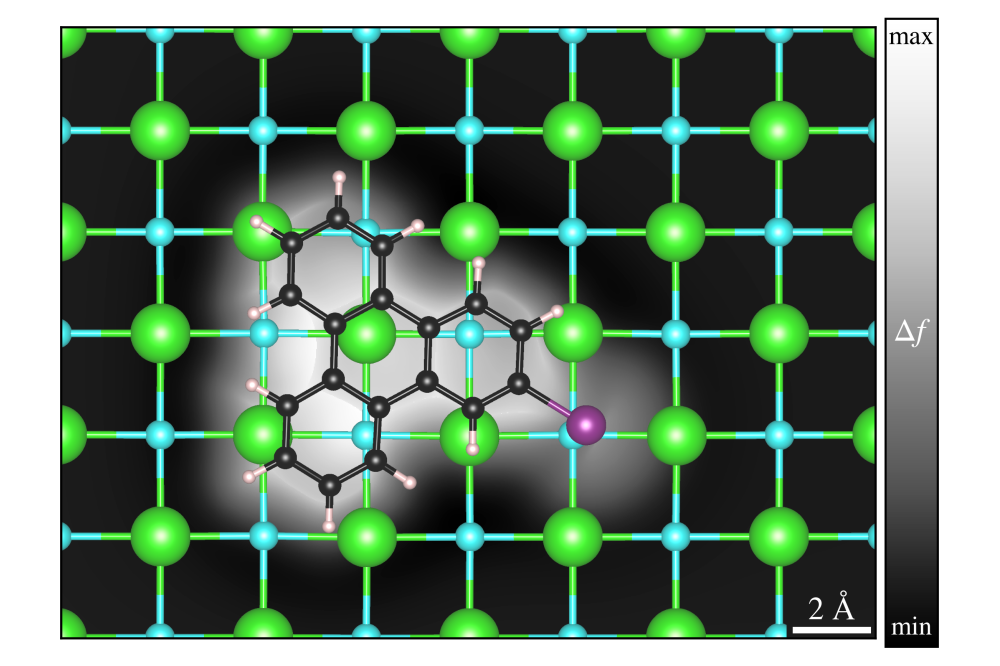
Simone Sanna_Principles of On-Surface Synthesis_Figure1Figure 1: Top view of the simulated adsorption geometry and AFM image of 2-iodotriphenylene on a NaCl monolayer. Na in light blue, Cl in green, H in white, I in violet and C in black.
Simone SannaEinleitung
In the past centuries chemists have established a solid knowledge for the synthesis of molecules in solution. With the precise knowledge of reaction paths, very high degree of sophistication for assembling chemical compounds has been achieved. Although this allows to produce large quantities of molecules, an atom-by-atom control of the produced molecules is not possible. The latter can be reached by on surface synthesis, instead. The in-depth knowledge about on-surface reaction mechanisms makes the tailor-made design of covalently bonded organic frameworks possible, which are used in applications such as graphene nanoribbons or quantum structures for nanoelectronic or -optical devices. In order to understand the underlying molecular mechanisms, we perform atomistic models of molecules and molecular reactions on different substrates. The outcome of experimental imaging experimental techniques such as AFM or STM are modeled as well.
Methoden
Our typical approach is based on atomistic calculations within the DFT for different exchange-correlation functionals as implemented in the plane wave codes VASP [1], Quantum Espresso [2], and Abinit [3]. These codes allow for very efficient calculations, especially in conjunction with the projector-augmented wave (PAW) method [4]. Structural relaxations as well as electronic structure calculations within the IPA for very large systems up to 1500 atoms have been realized with this approach by the applicant, using the VASP software package [5]. The latter is a computer program for first principles atomic scale materials modelling, e.g. electronic structure calculations and quantum-mechanical molecular dynamics. It offers efficient iterative matrix diagonalization techniques and highly customizable parallelization schemes. In particular, parallelization over bands, parallelization over plane wave coefficients, as well as parallelization over k-points can be used at the same time on massively parallel systems in order to obtain high computational efficiency. STM images are simulated in the Tersoff-Hamann approximation by the knowledge of the electronic structure, while AFM images are obtained by means of the particle-probe approach [6].
Ergebnisse
After thoroughly investigating the substrates [7], our calculations have contributed to identify the stable adsorption configurations of molecules such as dibromopyrene and 2-iodotriphenylene (see Figure 1) on sodium chloride coated copper substrates. On the basis of the DFT calculated geometries and wavefunctions, simulated STM and AFM images can be generated. This allows to interpret the experimental results and explain the contrast mechanisms [8]. In the case of dibromopyrene adsorbed on a NaCl monolayer deposited on a Cu(111) surface, the molecule is adsorbed with the two Br atoms close to Na atoms, resulting in an angle of ca. 19° between the molecular axis and the substrate [110] direction. The appearance of the AFM images strongly depend on the tip height above the substrate. Below 3.7 Å the inner part of the molecule is brighter, above 3.7 Å the inner part of the molecule is darker and the main contribution to the contrast mechanism is due to the halogen atoms [9].
Diskussion
Despite the recent progress in our understanding of the on-surface synthesis, many aspects have still to be investigated. Fundamental questions regarding the prerequisites for high reaction rates such as surface mobility and eventually surface defects are currently explored. The role of the substrate and the energetics of different reaction mechanisms are in the focus of the forthcoming investigations. Starting with the structural data of the single molecules, our project provides geometries and energetics of the possible reaction pathways of the investigated systems, thus helping the interpretation of the corresponding measurements and inspiring optimized on-surface synthesis.