Interatomic Potentials for Li-Si-O and Si-O-C
 Linus Erhard_Interatomic Potentials for Li-Si-O and Si-O-C_Figure1
Linus Erhard_Interatomic Potentials for Li-Si-O and Si-O-C_Figure1Figure 1: Atomistic model of silicon monoxide generated by an ACE potential for SiO.
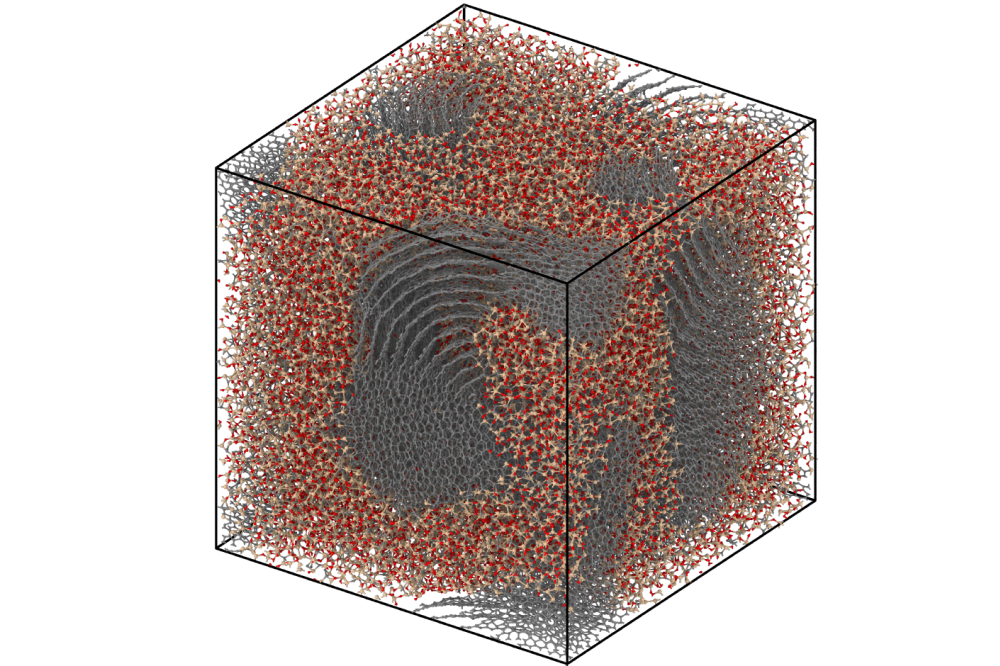 Linus Erhard_Interatomic Potentials for Li-Si-O and Si-O-C_Figure2
Linus Erhard_Interatomic Potentials for Li-Si-O and Si-O-C_Figure2Figure 2: SiOC structural model created by a melt-quench simulation using the ACE potential for SiOC.
Einleitung
To understand and study complex materials at the atomic level, it is essential to be able to calculate forces and energies for their atomic configurations. Density functional theory can be used to accurately calculate these properties, but these calculations are computationally expensive and subject to time and length constraints. Machine learning potentials overcome this problem and make it possible to simulate systems with millions of atoms for nanoseconds. In this project, we were interested in comparing several of these machine learning interatomic potentials. In addition, we analysed the highly complex SiO system using an existing machine learning potential and were interested in extending this machine learning potential to other systems. Powerful computing resources are essential for the high-throughput density functional theory calculations for the training data as well as the large-scale molecular dynamics simulation with the machine-learning interatomic potentials.
Methoden
As input data for the machine learning potentials, we use training data labelled with energies and forces from density functional theory. There are different types of machine learning potentials, depending on the machine learning method used and the descriptor of the atomic environment. In our case, we tested the Gaussian approximation potentials, the moment tensor potential and the atomic cluster expansion.
Ergebnisse
1. Benchmarking of different machine-learning interatomic potential approaches
For atomistic simulations, it is essential that the interatomic potential used is computationally efficient and accurate. There are now many types of machine-learning interatomic potential available. To find out which of these potentials offers the best performance for our system, we compare atomic cluster expansion, moment tensor potentials and Gaussian approximation potentials. We found that for the SiO system, the non-linear Atomic Cluster Expansion provides the best trade-off between accuracy and speed.
2. Generation of large scale silicon monoxide structures
The structure of silicon monoxide has long been controversial. Experiments have shown that it is a mixture of amorphous silicon and silicon dioxide. However, with only nanometre-sized grains. Until now, an atomistic description has not been possible because an exact description of the forces and energies on the required length scales was missing. We are now able to use a machine learning interatomic potentials to create structural models of silicon monoxide that are in excellent agreement with the experiment (see Fig. 1).
3. Construction of a Si-O-C database
Silicon oxycarbides are interesting for a wide range of applications. They can be used for
structural applications as well as battery material in lithium batteries. We have extended
the existing potential for SiO to carbon and significantly increased the database of
density functional theory data. With this new potential, we were able to create structures
containing phase-separated carbon and silicon dioxide as well as silicon carbide (see Fig. 2).
4. Construction of a Li-Si-O database
Unfortunately, due to time constraints, it was not possible to complete this part of the
project within the project duration
Diskussion
Through machine learning interatomic potentials trained on a large amount of density functional theory data, we can gain new insights into materials. We have demonstrated this by generating realistic nanostructures of SiO and SiOC. In the future, these machinelearning interatomic potentials could be used to investigate a wide range of other problems in SiO and SiOC to improve our understanding of these systems. Future work is also planned to extend the machine learning interatomic potential models to lithium.