Multiscale Modelling of Cement-Based Materials II
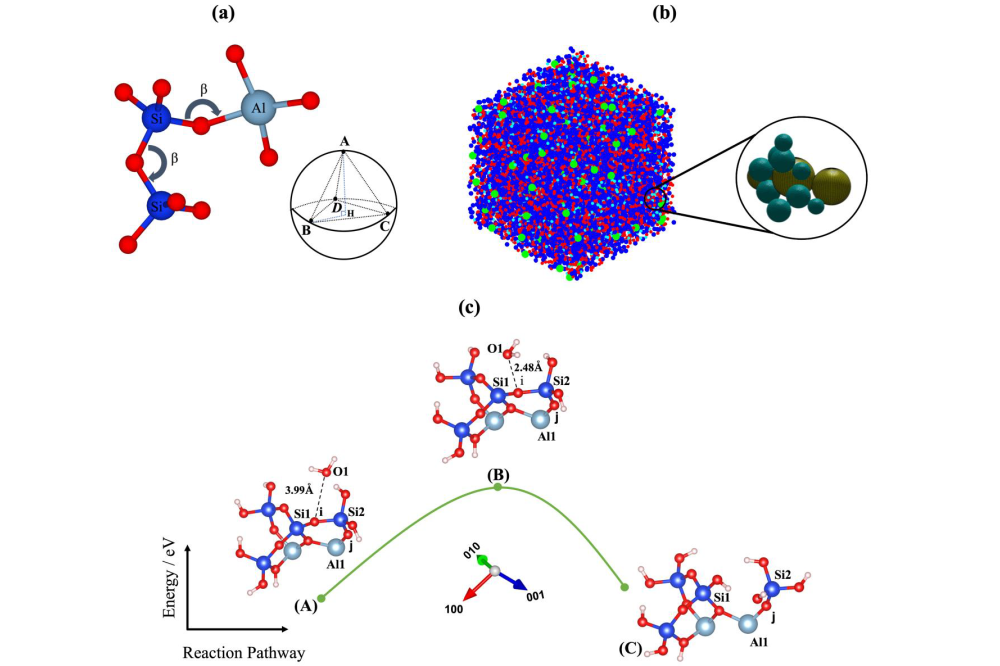 Mohammadreza Izadifar_Multiscale Modelling of Cement-Based Materials II_Figure1Figure 1: (a) Formation of Si-O-Si and Si-O-Al bonds which respect the β angle and tetrahedral geometrical constraints. (b) The final status of the simulation with particles, clusters, and pore distributions arrangements. (c) Activation energy (EB – EA) and optimized geometric structures of the hydrolysis dissolution reaction: reactant (A), transition state (B) and products (C).
Mohammadreza Izadifar_Multiscale Modelling of Cement-Based Materials II_Figure1Figure 1: (a) Formation of Si-O-Si and Si-O-Al bonds which respect the β angle and tetrahedral geometrical constraints. (b) The final status of the simulation with particles, clusters, and pore distributions arrangements. (c) Activation energy (EB – EA) and optimized geometric structures of the hydrolysis dissolution reaction: reactant (A), transition state (B) and products (C).Einleitung
The project aims to develop a bridging model that connects the nanoscale to the upscaled levels for understanding the chemical reaction mechanisms in cement-based and geopolymer-based materials. This involves utilizing atomistic modeling techniques such as ab-initio (DFT), molecular dynamics (using meta-MD and ReaxFF) to gain insights into the underlying processes. Upscaled modeling approaches like atomistic kinetic Monte Carlo (kMC) and Coarse-Grained Metropolis Monte Carlo are used to quantify and validate reaction rates and surface interface chemistry. Additionally, mesoscale methods such as Finite Element and Finite Difference, along with phase-field modeling, are employed to study reactive transport mechanisms under different environmental conditions.
Methoden
Atomistic modeling approaches, including ab initio Vienna simulation package (VASP) for self-consistent computations and equilibrium DFT for different configurations as well as Molecular Dynamics (MD, ReaxFF) via LAMMPS package, are employed in these projects. For upscaled coarse-graining modeling, kinetic Monte Carlo (kMC) using an in-house MATLAB code and off-lattice coarse-grained Monte Carlo (CGMC) methods are utilized.
Ergebnisse
In [1], MD calculations enabled to obtain the activation energies for the identified atomistic events, which can be used to calculate the atomistic reaction rates employing a transition state theory. In [2], the calculated atomistic forward reaction rates based on [1] were computed for 21 different event scenarios. To observe the effect of crystal defects on the dissolution rates, a comparison was drawn with the ideal crystal morphology. In [3], 3D off-lattice coarse-grained Monte Carlo (CGMC) model has been implemented for studying the nucleation of alkaline aluminosilicate gel (Figure 1A) for the silicate-activated system. Therein, typical geopolymer monomer species are coarse-grained as different particle types, to compute the gel structure evolution with geopolymerisation reaction advancement. In [4], the activation energy for computing atomistic reaction rates was determined for the dissolution of silicate tetrahedra from metakaolin precursors under far-from-equilibrium conditions. This computation was based on the transition-state theory using the DFT computational approach.
Diskussion
It has been observed that certain facets of belite clinker are found more reactive during the initial hydration stage, while less reactive compared to the alite clinker mineral. This is due to the crystal structure, absence of free oxygen and higher number of silicate crystal sites [1]. Moreover, the layer-by-layer dissolution mechanism observed could also explaine the slow reactivity of belite clinker [2], as well as some alite crystal planes. The on-lattice CGMC model developed in [3], was in better agreement with the Gaussian distribution and the measurement results than the off-lattice approach. The results obtained from [3-5] provide missing inputs for further upscaling investigations. These investigations aim to establish a comprehensive database for coarse-grained (and/or kinetic) Monte Carlo simulation, considering various input chemistries and for calculating the materials mechanical properties.