Rational Design of Narrow-Hysteresis Materials by Tailoring Magnetoelastic Interactions II
 Olga N. Miroshkina_Rational Design of Narrow-Hysteresis Materials by Tailoring Magnetoelastic Interactions II_Figure1
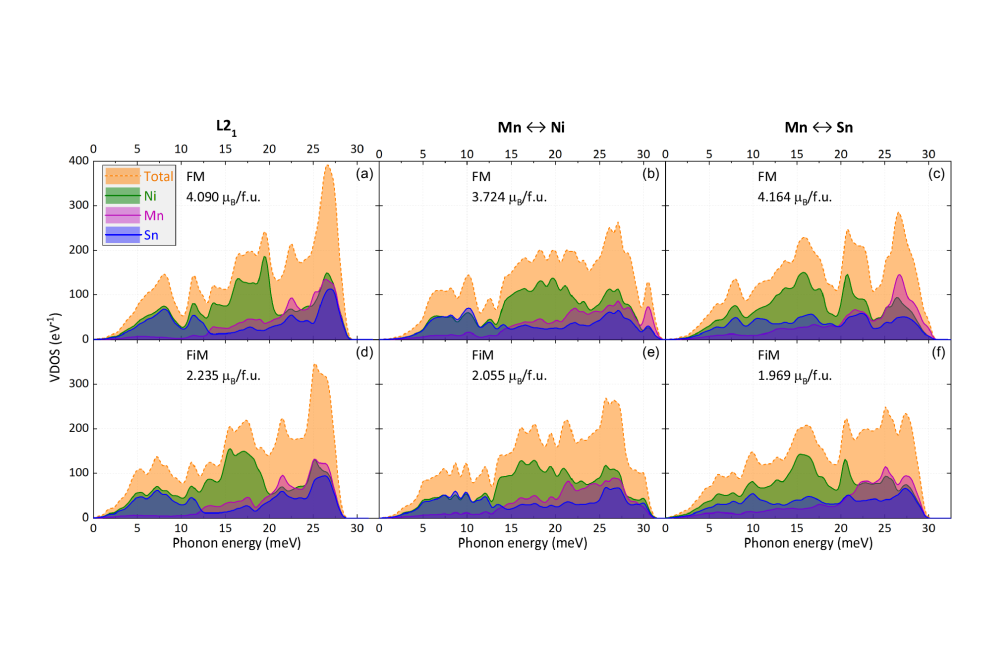
Olga N. Miroshkina_Rational Design of Narrow-Hysteresis Materials by Tailoring Magnetoelastic Interactions II_Figure1Figure 1: Calculated total and element-resolved VDOS for the ideal L21 structure (a), (d) and systems with antisite disorder introduced by swapping of Mn and Ni atoms (b), (e) and Mn and Sn atoms (c), (f). Gaussian smearing with a width of 0.0725 THz is used for better visibility. Results for the ferromagnetic (FM) and ferrimagnetic (FiM) configurations are presented in parts (a)–(c) and (d)–(f), respectively. The magnetic moments of each structure (in μB/f.u.) are given in the figures. Copyrighted by APS. Permission for reproduction in technical report has been granted. Published in Phys. Rev. B 106, 214302 (2022).
Einleitung
The current project aims to reveal the relevant intrinsic mechanisms responsible for the coupling of the lattice and magnetic degrees of freedom, providing insight into their contribution to the first-order nature of the magnetic phase transition and hysteresis in state-of-the-art magnetocaloric materials. We carry out the first-principles study of the electronic structure and calculate the total energy, magnetic properties, and electronic and vibrational density of states as a function of the macroscopic state variables (magnetization, volume, and strain). From these data, thermodynamic quantities such as free energy or entropy can be derived, which allow getting information about the contribution of specific elements or particular degrees of freedom. This, in turn, enables us to characterize the free energy barriers with respect to the coupling between electronic, lattice, and magnetic degrees of freedom. In the end, we aim to predict novel highly-efficient multifunctional materials with narrow hysteresis and provide an insight into the intrinsic mechanisms.
Methoden
Ergebnisse
During last year, we calculated the energy phase diagram of the Ni-Mn-(Sn,In) Heusler alloys. We compared the energy of ferrimagnetic L10, ferromagnetic L21, and twinning ferrimagnetic structures of Ni8Mn5In3 and Ni8Mn5Sn3. The most attention, however, was paid to the stoichiometric Ni2MnSn Heusler alloy focusing on its magnetic and vibrational properties. We calculated total and partial vibrational densities of state (VDOS) of the ideal L21 structure and compared Sn-VDOS with the experimental results of our collaborators. We found a nice agreement in the low-frequency range, while the agreement was less obvious in the high frequencies. To find the reason, we calculated VDOS for structures with two variants of antisite and magnetic disorder. Using results of phonon calculations performed on Lichtenberg HPC, we associate the peaks in the VDOS with particular features in the element-resolved phonon dispersion of L21 ordered Ni2MnSn.
Diskussion
The good agreement between theory and experiment in the low-energy region provides the evidence that the inversion of optical modes at Γ involving the displacement of Ni and the heavier main group element atoms, which was predicted previously for other Ni-Mn-based Heusler compounds, is also a characteristic property of Ni2MnSn. Introducing different types of magnetic and antisite disorder in our calculations results in a distinctive redistribution and broadening of the Sn-VDOS. Mössbauer spectroscopy at low temperatures confirms this suggestion revealing the presence of several inequivalent Sn sites, which indicates the presence of a considerable amount of site-disorder and other defects (such as, e. g., antiphase boundaries) in the sample. The comparison of the site-resolved VDOS obtained with different kinds of site-disorder realized in our modeling shows that the large peaks at high energies are indeed susceptible to changes in the electronic structure of the environment. These may arise, e. g., from chemical or magnetic disorder or particular modeling of exchange and correlation on the transition metal sites, which explains the remaining differences between the experimental and calculated Sn-VDOS. We showed that the particular deviations between theory and experiment can be discussed in terms of structural and magnetic defects. The good agreement with experimental data documents the predictive power of DFT also for the vibrational properties of Heusler compounds with main group elements from the fourth row.