Multiscale Modelling of Cement-Based Materials
 Neven Ukrainczyk_Multiscale Modelling of Cement-Based Materials_Figure1
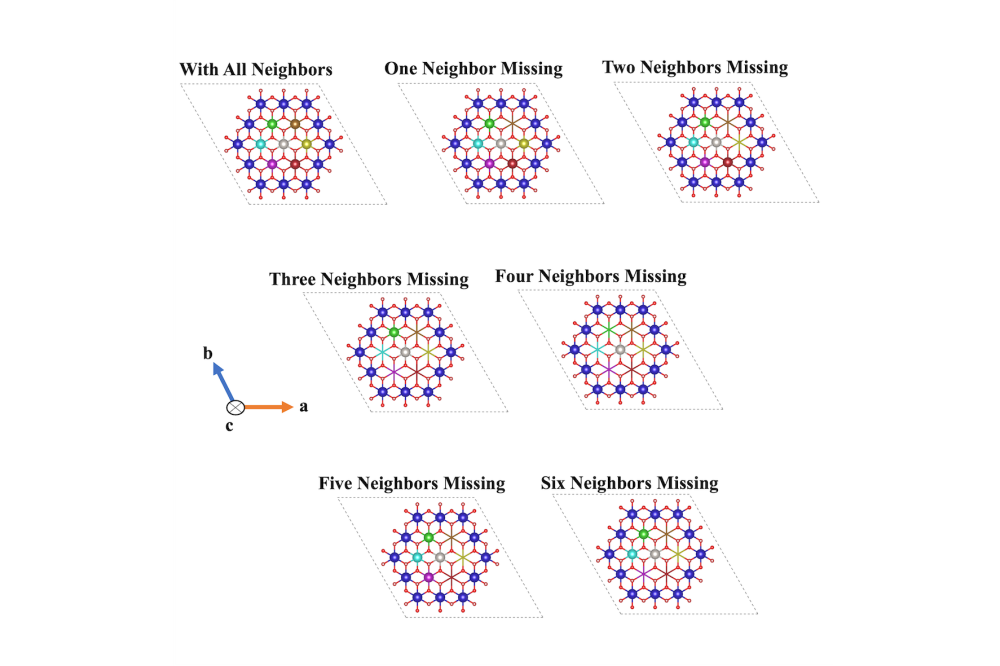
Neven Ukrainczyk_Multiscale Modelling of Cement-Based Materials_Figure1Figure 1: Atomistic dissolution scenarios of hexagonally oriented central Ca (light gray) from (001) crystal facet of Portlandite, proposed to feed the KMC upscaling approach.
Neven Ukrainczyk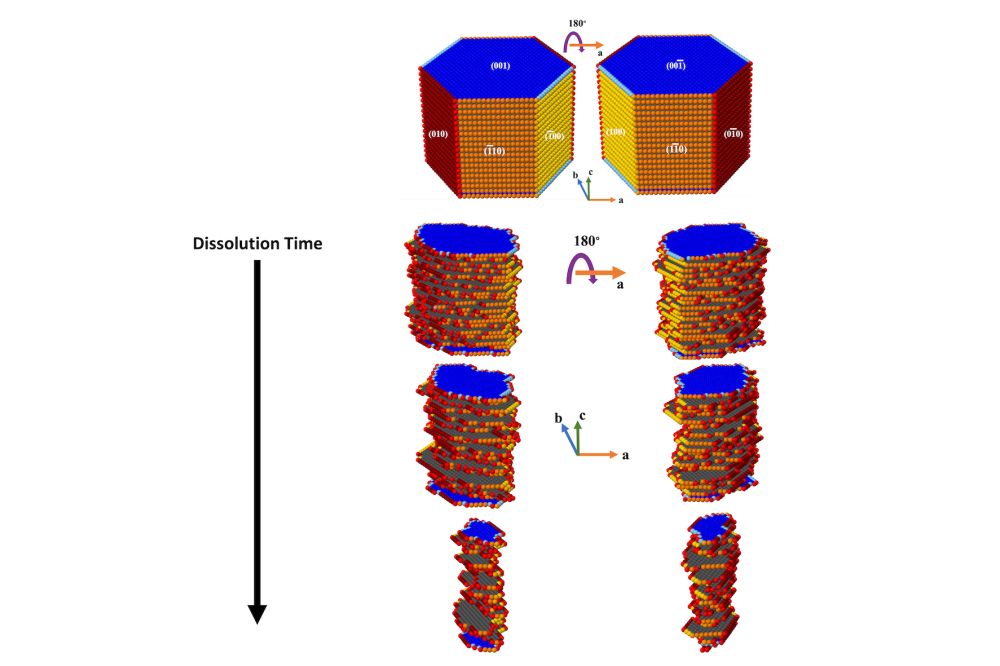 Neven Ukrainczyk_Multiscale Modelling of Cement-Based Materials_Figure2
Neven Ukrainczyk_Multiscale Modelling of Cement-Based Materials_Figure2Figure 2: Evolution of Portlandite crystal morphology during an atom-by-atom site dissolution KMC simulation. For better visualization each facet is shown with different colors, while atoms with less neighbors have lighter color tone.
Neven UkrainczykEinleitung
Overall objective of this project is to develop an elementary physical/chemical bridging model for the chemical reaction mechanisms of cement-(and geopolymer)-based materials that connects the nanoscale to the upscaled levels. The approach starts with an in-depth understanding by various atomistic modelling approaches (DFT, meta-MD, ReaxFF). Upscaled modeling approaches are based on kinetic Monte Carlo (kMC) and other particle based (Coarse Grained Metropolis Monte Carlo) approaches. The upscaled model will quantify and validate the reaction rates and surface interface chemistry, as an essential process which later has to be considered by models attempting to capture fundamentals of building materials properties. At the meso-scale, Finite Element and Finite difference (and phase-field) methods will be used for reactive transport mechanisms, e.g. in order to simulate materials behavior under different environmental conditions and at different stages.
Methoden
Various atomistic modelling approaches are used. Vienna ab initio simulation package (VASP) was installed 2021 and will be used as a mostly. Other methods are Molecular Dynamics (MD, ReaxFF) vai LAMMPS package and Upscaled coarse-graining modeling approaches are based on kinetic Monte Carlo (kMC, as in-house code developed in MATLAB) and other coarse-grained Metropolis Monte Carlo approaches (in-house developing MATLAB code). At the meso-scale, Finite Element phase-field methods are used (MOOSE).
Ergebnisse
The comparison between quantum chemistry (DFT) [1] and molecular dynamics (ReaxFF-metaD) results [2] showed very good qualitative agreement for atomistic activation energies, capturing same trends as a function of crystal neighbor configurations. For KMC, 16 different atomistic scenarios for Ca dissolution were considered (Fig. 1) depending on the existing neighbors for portlandit crystal facets. This allowed for KMC upscaling of the atomistic dissolution rates of the different scenarios into a mesoscale rate and enabled the visualization of the evolution of crystal morphologies during the dissolution process (Fig. 2). Results of the upscaled KMC simulations demonstrate that dissolution process initially takes place from edges, sides, and preferentially from the 010 crystal facet. In Yang et al. publication [3], a novel phase-field (PF) based model for self-healing of cementitious materials was developed. The model effectively captured the evolution of dissolution and precipitation of meso-scale crack interfaces controlled by diffusion driven mechanism of solute concentration profiles.
Diskussion
The atomistic KMC results [1] showed that portlandite upscaled dissolution rates between each crystal facet resulted in a huge, 16 orders of magnitude difference, reflecting the importance of crystallographic orientation of the exposed facets. The steady-state dissolution rate for the most reactive facet (010) was found to be 1.0443 mol/(s cm2), while the less reactive facet (100) had a rate of 0.31 × 10-16 mol/(s cm2). These results are important for a general understanding of the cement hydration and other chemical processes with portlandite. The PF simulations [3] with the derived interfacial mobility parameter showed a consistent agreement with the experimental results. The interfacial growth kinetics in terms of minerals dissolution and precipitation during the self-healing of cementitious materials was successfully simulated in 2D. Comparison with experimental results showed that the PF model was able to provide good qualitative predictions of the morphological and geometric details of the interfacial process.