Rational Design of Narrow-Hysteresis Materials by Tailoring Magnetoelastic Interactions
 Miroshkina, Olga_Rational Design of Narrow-Hysteresis Materials by Tailoring Magnetoelastic Interactions_Figure1
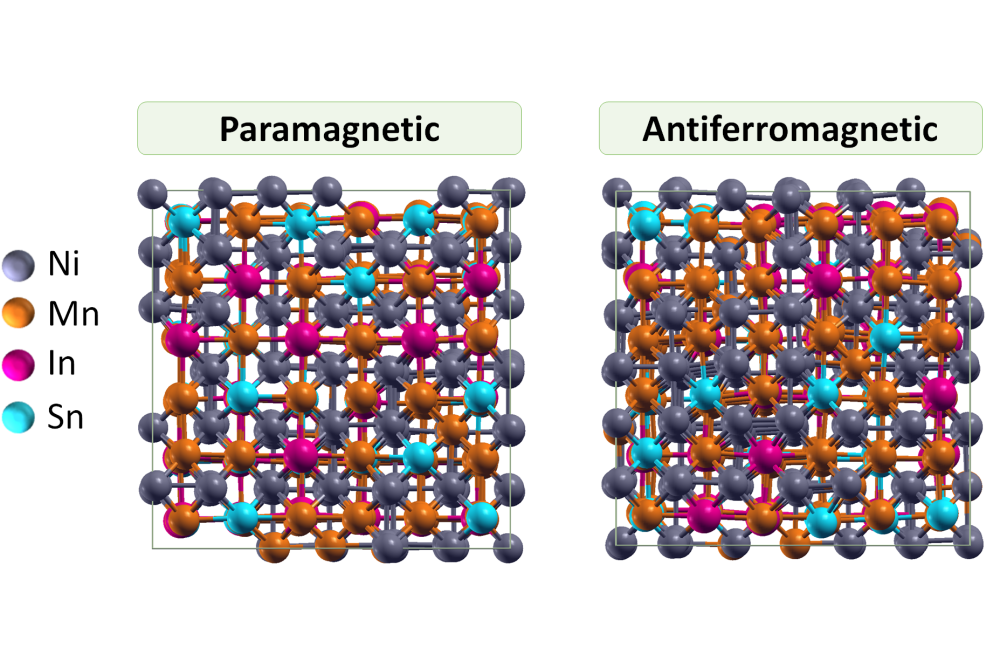
Miroshkina, Olga_Rational Design of Narrow-Hysteresis Materials by Tailoring Magnetoelastic Interactions_Figure1Figure 1: Top view of the relaxed crystal cubic crystal structures of Ni48Mn36In16–xSnx in the PM and AFM phase. The latter exhibits substantially stronger relaxations.
Olga Miroshkina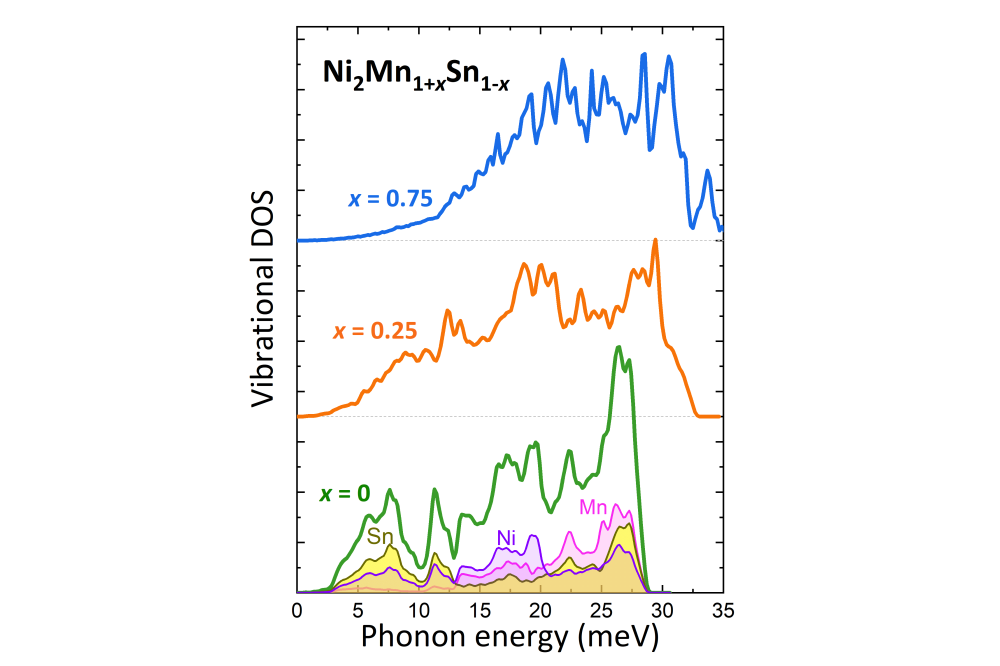 Miroshkina, Olga_Rational Design of Narrow-Hysteresis Materials by Tailoring Magnetoelastic Interactions_Figure2
Miroshkina, Olga_Rational Design of Narrow-Hysteresis Materials by Tailoring Magnetoelastic Interactions_Figure2Figure 3: Vibrational densities of states of Ni2Mn1+xSn1–x (x = 0;0:25;0:75) in ferromagnetic configuration. The lower panel also shows the element-resolved decomposition of the VDOS.
Olga MiroshkinaEinleitung
One important aim of modern energy-conserving technologies is magnetic cooling at room temperature. This environmentally friendly and highly efficient technology is based on the magnetocaloric effect (MCE), which can be explained as a thermal response of magnetic material on the change of the external magnetic field. Despite the intensive studies, the way from the prototypes to the effective systems in mass production is not fully performed, since the optimal combination of the large MCE and narrow hysteresis in one material is difficult to achieve. One of the main trouble is the transition-hysteresis, which is associated with the dissipation energy losses, and now it is in the focus of the materials research community. The goal of current project is a comprehensive theoretical study of the free energy barriers leading to non-reversible transition behavior in the multifunctional materials, which help to make a significant step into the understanding of the intrinsic mechanisms of hysteresis.
Methoden
The major part of the calculations was performed in the framework of DFT as implemented in the Vienna ab initio simulation package (VASP). The code uses a plane-wave basis set to describe the wavefunctions of the valence electrons and treats the interaction with the core electrons within the Projector Augmented-Wave (PAW) approach. Exchange-correlation energy was treated in the framework of the semi-local generalized gradient approximation, which depends on the local electron density and its gradients, in the Perdew-Burke-Ernzerhof (PBE) formulation and its variation PBEsol. Phonon calculations were performed in the framework of the direct (force-constant) method. VASP was used for the calculation of Hellmann-Feynman forces, while for postprocessing and the selection of the atomic displacements we used Phonopy.
Ergebnisse
In the first step, we modeled the energy phase diagrams of several magnetocaloric Heusler systems including Ni-Mn-(In,Sn) and so-called all-d Ni-Mn-Ti. Here, we have studied the compositions of different stoichiometry Ni2Mn1+xZ1–x with concentration steps of x = 0:25. Ferromagnetic (FM) and ferrimagnetic ordering of the Mn-atoms in the 4a- and 4b-sublattices were modeled. The latter is characterized by the antiparallel (AFM) orientation of Mn-excess magnetic moments with respect to the other Ni and Mn atoms. We determined the energetically favorable magnetic state in the austenitic and – if possible – martensitic phases. For favorable structures, we obtained the phonon spectrum, VDOS, and thermal properties such as free energy, heat capacity, and entropy were obtained.
Comprehensive first-principles investigation is performed for the magnetocaloric Ni-Mn-(In,Sn) Heusler compounds, which are under investigation by our experimental partners at the university of Parma, in particular Dr. Francesco Cugini. We considered different magnetic configurations (FM, ferrimagnetic and paramagnetic (PM) with full magnetic disorder) in a 432-atom supercell and calculated the total energies after full relaxation of the atomic positions. We found strong structural relaxation in the PM and especially in the ferrimagnetic (here denoted as AFM) cases.
Diskussion
We compared the DFT total energy of Ni-Mn-(In,Sn) with experiment and magnetic exchange constants calculated with multiple scattering theory. It was found that the nearest neighbor (NN) and next-NN direct exchange between the transition metals Ni and Mn is modified by the exchange of the main group elements In and Sn. This is fostered by a strong competition between FM Ni-Mn4a;4b and AFM Mn4a-Mn4b exchange such that increasing of valence electron number in the system caused by shifting from In to Sn forces a ferrimagnetic ground state. In combination with additional Mn-excess on the Ni8c sites, compositions with Sn content in excess of In develop a complex ground state with the frustrated interactions between the sublattices, which perfectly explains the experimental observations.
The calculated VDOS for ternary Ni-Mn-Sn alloys are compared with the experimental results of our collaborators. The group of Prof. Dr. Heiko Wende (University of Duisburg-Essen) measured Sn-VDOS on the samples prepared and characterized in the group of Prof. Oliver Gutfleisch (TU Darmstadt). We found, that our theoretical Sn-VDOS reproduces well the experiment for Ni50Mn35Sn15, while in the stoichiometric case we obtain an additional sharp peak at high frequencies, which is not observed in experiment. We suppose that this difference is associated with the ubiquitous residual disorder in the experimental sample, which need to be taken into account in the calculations as well. Respective calculations for structurally disordered systems are planned for the coming period.