Defect Properties of Lead-Free Antiferroelectrics for Energy Storage Applications
 Defect Properties of Lead-Free Antiferroelectrics for Energy Storage Applications1
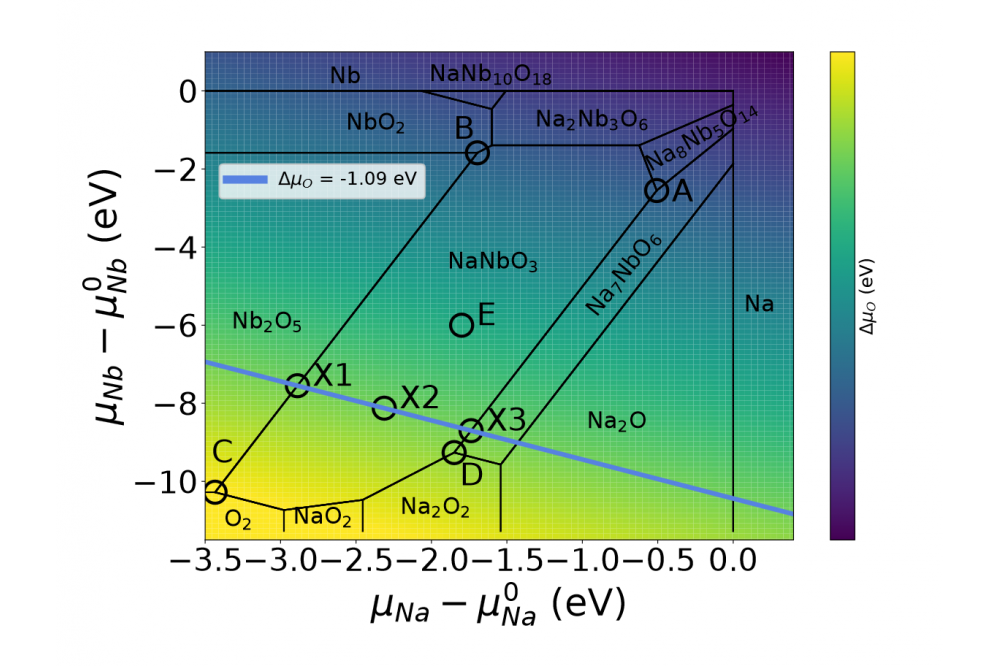
Defect Properties of Lead-Free Antiferroelectrics for Energy Storage Applications1Figure 1: Stability diagram of the ternary system Na-Nb-O with HSE xc-functional. The defect formation energies are discussed in terms of the chemical potentials at the points from A to E.
Lorenzo Villa Defect Properties of Lead-Free Antiferroelectrics for Energy Storage Applications2
Defect Properties of Lead-Free Antiferroelectrics for Energy Storage Applications2Figure 2: Defect formation energies as a function of the Fermi level position for representative thermodynamic conditions of NN (shown in Figure 1).
Lorenzo VillaEinleitung
Dielectric capacitors are considered to be promising candidates for energy storage applications in high power electronics, due to their advantages of fast charge-discharge capability and high power density compared to batteries or fuel cells. Among the currently available dielectric capacitors, antiferroelectric (AFE) materials are outperforming ferroelectrics (FE) and linear dielectrics, because of the unique double polarization vs. electric field (P-E) loops. However, their practical application in high energy storage devices is severely restricted by the low energy density and energy efficiency. Thus, overcoming these drawbacks has the highest priority in recent dielectric energy storage materials researches. In this class of materials, AgNbO3 and NaNbO3 ceramics were actively investigated and reported due to the high energy storage performance and environmentally friendly characters. However, the energy density and energy efficiencies are urgently needed to be further optimized in order to reduce the size of the device and limit energy losses. In this context, ab-initio simulations can provide new information in order to understand the atomistic features of these systems and to discover the best way to optimize their energy storage properties.
Methoden
All calculations were performed based on density functional theory (DFT) using the Vienna ab- initio simulation package (VASP). The electronic wave functions were described using the projector augmented-wave method, which includes both valence and core states. The exchange-correlation effects were treated using the Perdew-Burke-Ernzerhof formalism of the generalized-gradient approximation (GGA), the Dudarev version of GGA+U and the HSE06 hybrid functional.
Ergebnisse
The formation energies, equilibrium defect transition levels and doping behaviour of intrinsic defects in cubic NaNbO3 have been determined by means of Density Functional Theory (DFT) (submitted manuscript). As shown in Figure 2 (with reference chemical potentials in Figure 1), Nb vacancies are found to have high formation energies, while Na and O vacancies are calculated to have relatively small formation energies. The calculation of the equilibrium electron chemical potential shows that in Na-poor/O-rich conditions the semiconductor tends more towards being p-type, while in all other conditions the electron chemical potential is closer to the conduction band, resulting in an n-type semiconductor. Finally, the binding energy of the complex formed by two Na vacancies and one O vacancy is found to be negative, which indicates that defect complexes might be present in relatively large concentrations. The electron chemical potential calculation shows that intrinsic AN is a p-type semiconductor. Moreover, the calculations of the Mn substitutional defect show that Mn is stable on the Nb-site, with stable charge states of -3,+1 and +2. This indicates that MnNb can be a donor or acceptor based on the position of the electron chemical potential. This study confirms that Mn-substitution in AgNbO3 is a complex mechanism, which can only be described by a thermodynamic model that relies on computed data and not on a priori assumptions.
Diskussion
We have investigated the defect equilibrium in NaNbO3 and AgNbO3, which are candidates for lead-free antiferroelectrics capacitors. In both systems vacancies in the A-site (Ag and Na) and oxygen vacancies have been found to be present in relatively large concentrations, which can contribute to shift considerably the electron chemical potential of the material also in intrinsic conditions. Moreover, the binding energy of the complex formed by two Na vacancies and one O vacancy in NN is found to be negative, which indicates that defect complexes might be present in relatively large concentrations and therefore affect the P-E loops, where previously only dopant-vacancy complexes have been proposed as an influencing factor.