Ab-initio Modeling of Battery Materials
 Sadwoski, Marcel,Ab-initio Modeling of Battery Materials
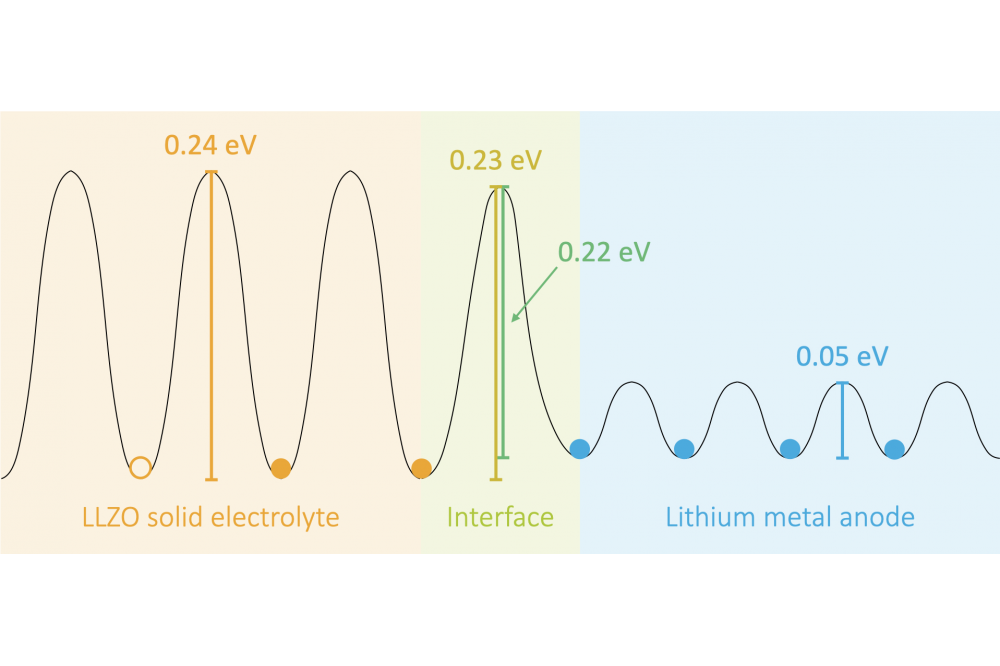
Sadwoski, Marcel,Ab-initio Modeling of Battery MaterialsFigure 1: Schematic energy landscape for a lithium vacancy travelling from the LLZO electrolyte over the LLZO|Li metal interface into the metallic lithium.
Marcel SadowskiEinleitung
Improved batteries are needed to meet increasing energy demands worldwide (e.g., portable and medical devices, e-mobility, renewable energies). One approach to develop such energy storage systems relies on so-called all solid-state batteries which are able to deliver high energy and power densities and simultaneously ensure safety due to the absence of flammable liquid components. To this end solid electrolytes (SE) with low electronic but high ionic conductivity need to be developed that furthermore comply with the electrochemical and mechanical conditions at the interfaces towards the electrodes. Throughout several research projects on the Lichtenberg Cluster we have therefore applied computational methods on the atomic level to investigate the interplay between structure, thermodynamics and transport properties of various SE in order to accelerate the materials development. Our most recent findings are highlighted in the following and relate to the influence of Br-/S2- anion site-disorder on the Li transport properties in Li6PS5Br and the interface between Li6.25Al0.25La3Zr2O12 (LLZO) and metallic Li.
Methoden
Atomistic simulations within the density functional theory (DFT) have been performed using the Vienna ab-initio simulation package (VASP) with projector augmented-wave pseudopotentials and the PBE exchange-correlation functional. For the respective materials under investigation calculation parameters like the cutoff energy of the plane-waves and the k-point sampling have been converged prior to the production steps. Initial atomic structures for crystalline materials were obtained from literature or collaboration partners. Used amorphous structures have been generated in the past using a simulated melt-quenching scheme in ab-initio molecular dynamics simulations (AIMD) [1]. AIMD simulations were also used to study transport properties by inspection of the Li trajectories, their mean-square displacements (MSD) and counting of local jump events.
Ergebnisse
For the argyrodite-type Li6PS5Br we continued our previous studies [2] and could consolidate that site-disorder on the Br-/S2- anion sublattice has an crucial influence on the Li diffusivity [3]. Without site-disorder the S2- sites are completely surrounded by Li in a cage-like manner and diffusion mainly occurs only within the Li cages. With increasing site-disorder the strict cage structure is disrupted and the Li ions becomes more homogeneously distributed within the material. Calculated Li density distributions match the experimental observations clearly indicating that the anion site-disorder is responsible for the improved transport properties. Furthermore, we showed that at low degrees of site-disorder the transport mechanism can be described as a hopping of Li interstitials. Because high site-disorders are observed at elevated temperatures we tried to involve contributions to the configurational entropy in order to predict the equilibrium site-disorder as function of temperature. However, we could show that the contribution of the anion site-disorder alone is insufficient [3].
In terms of the garnet-type LLZO we constructed explicit interfaces towards Li metal and investigated the interface dynamics at various temperatures. First, the calculations show that the interface is indeed stable even up to temperatures above 1000 K. Second, diffusion across the interface has been observed. More detailed analyses involving AIMD simulations, counting of Li jumps and nudged elastic band calculations revealed that a jump across the interface has nearly the same migration barrier as jumps within the LLZO bulk (see Figure 1). Therefore, the resistance increase, often observed in experiments during cycling, most probably originates from the formation of pores and contact loss at the interface.
Ausblick
Clearly, the bulk properties of materials are key parameters to assess whether a material is suitable for an application. In all-solid-state batteries, however, also the interfacial properties are becoming more and more of an issue. Therefore, interfaces between different materials but also internal interfaces such as grain boundaries will be the topic of our future studies.