Investigation of Vacancy Diffusion in Crystalline LIPON Compounds by Means of Density Functional Theory
 Henkel, Pascal; Investigation of vacancy diffusion in crystalline LIPON compounds by means of density-functional theory; Figure 1
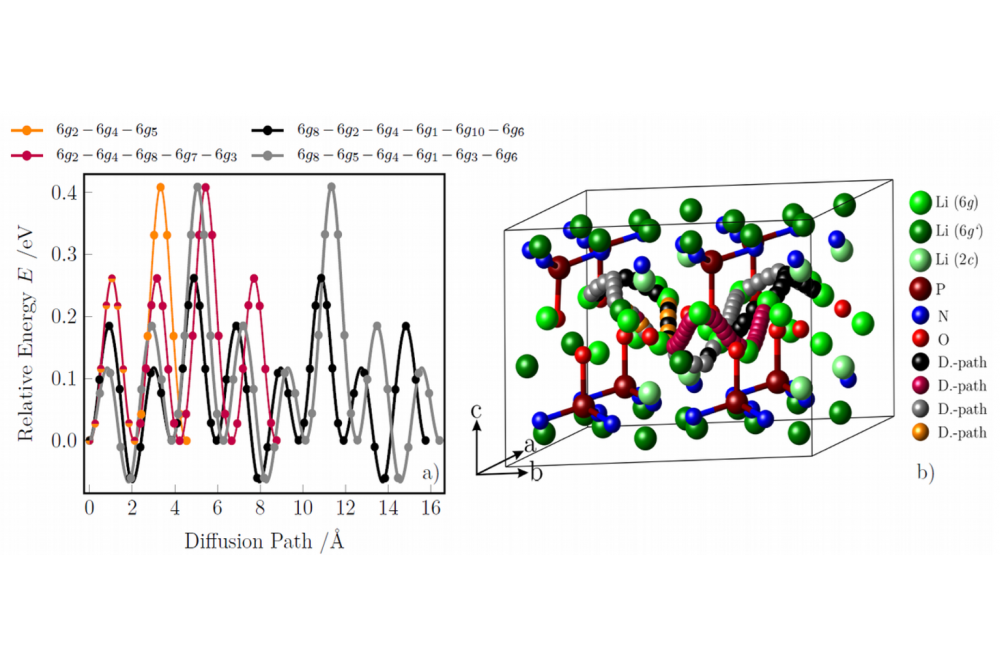
Henkel, Pascal; Investigation of vacancy diffusion in crystalline LIPON compounds by means of density-functional theory; Figure 1Figure 1: Diffusion path of lithium vacancy diffusion at the 6g Wyckoff position for the LIPON structure with isolated PON36- tetrahedrone. (0D-LIPON) a) energetic and b) graphical representation of the different diffusion paths [6].
Pascal Henkel Henkel, Pascal; Investigation of vacancy diffusion in crystalline LIPON compounds by means of density-functional theory; Figure 2
Henkel, Pascal; Investigation of vacancy diffusion in crystalline LIPON compounds by means of density-functional theory; Figure 2Figure 2: Diffusion path of the lithium vacancy diffusion along the c-axis for the LIPON structure with chain-linked PO2N22-tetrahedrone. (1D-LIPON) a) energetic and b) graphical representation of the different diffusion paths [6].
Pascal Henkel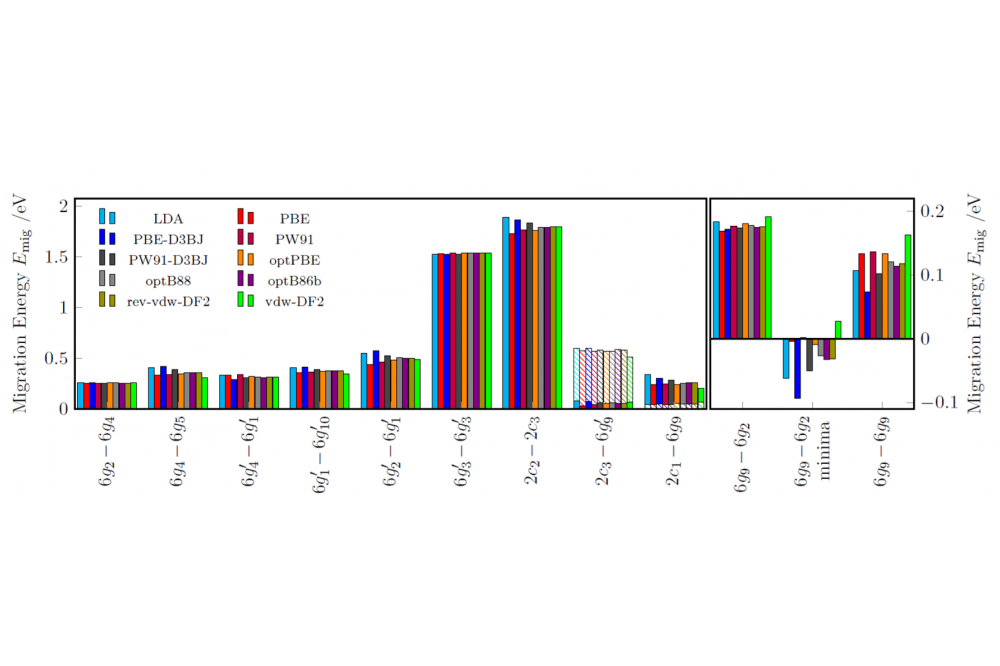 Henkel, Pascal; Investigation of vacancy diffusion in crystalline LIPON compounds by means of density-functional theory; Figure 3
Henkel, Pascal; Investigation of vacancy diffusion in crystalline LIPON compounds by means of density-functional theory; Figure 3Figure 3: Influence of 11 different exchange-correlation-functionals on the diffusion barriers in LIPON (isolated PON3 -tetrahedrone structure). Forth (full filled) and back diffusion (stiffened fiiled) differ for the two pathways 2c3-6g'9 and 2c1-6g9 [7].
Pascal HenkelEinleitung
Due to an ever-increasing energy demand worldwide, new and efficient energy storage systems are needed more and more. Lithium-ion batteries have emerged as a promising storage medium and therefore have been the subject of intensive research for decades. The performance of such batteries is significantly dependent on the electrode-electrolyte interaction [1]. In this context, Lithium Phosphorus Oxynitride (LIPON) has proven to be a solid state electrolyte of great interest [2]. LIPON compounds have the stoichiometry LixPOyNz with x = 2y+3z-y. Due to the different POuN4-u (with u = 1-4) building blocks a variety of n dimensional structures are possible [3-5]. For a better undestanding of the influence of these n dimensional anionic bilding blocks on defect formation energies and lithium diffusion, we have investigated two crystalline structures [3,4].
Methoden
To determine the diffusion barriers and the defect formation energies of lithium vacancies of LIPON bulk phases, we applied density functional theory using VASP [5]. We not only investigated the diffusion barriers, but also the influence of different functionals and dispersion corrections.
Ergebnisse
We are presenting our density functional theory results for two selected crystalline LIPON structures. The first LIPON structure (see Fig. 1, 0D structure) consists of three energetically non-equivalent lithium positions, with the 6g Wyckoff position being the most energetically favourable (green), followed by the 2c-position (light green) with a relative energy of 0.30 eV and finally the 6g' position (dark green) with a relative energy of 0.82 eV. This structure is composed of PON3 acid building blocks. The deviating energies of the 6g and 6g' positions are due to the fact that the 6g' position is located neat the three nitrogen atoms, whereas the 6g' position is located at the O-tip of the PON3 tetrahedron. This means that vacancy diffusion processes can take place both in the ab-plane (on all three Wyckoff positions) and in c-direction between the Wyckoff positions. The diffusion within the 6g position is - with 0.27 eV - the energetically most favourable. In addition, it is demonstrated that the diffusion from a 6g' to a 6g position with a barrier of 0.01 eV is significantly more energetically favourable than the diffusion within the 6g' position (within the plane: 0.41 eV). The analogue is observed for the 2c position (within the plane: larger than 1.8 eV and from 2c to 6g: 0.04 eV). Therefore we have shown for this 0D LIPON structure that the vacancy diffusion takes place exclusively within the 6g position, whereas a diffusion along the c-axis from the 6g position is energetically not to be expected. In the second LIPON structure (see Fig. 2, 0D structure) the lithium ions exclusively occupy the 8b-Wyckoff position. This structure is composed of PO2N22- chain-linked building blocks. In this structure the vacancy diffusion is not determined by the Wyckoff positions, but by the orientation to the PO2N22- chains. It is shown that the diffusion perpendicular and vertical to the chains is energetically equal (with 0.62 eV). However, the diffusion parallel to the chain alignment (see Fig. 2 orange curve) with 0.41 eV is energetically the most favourable and so the preferred one. This diffusion takes place parallel to two stacked PO2N22- chains, whereas the diffusion parallel to one chain (see Fig. 2 black curve) is energetically unfavourable (larger than 1.6 meV)[6]. As shown, the microscopic structure and orientation of the anionic building blocks have a considerable influence for the diffusion. A comparison of 11 different functionals (with and without dispersion corrections and van-der Waals functionals) demonstrates that the choice of a suitable functional is important. Also, overall the functionals provide similar same results, differences can be observed on examining the different diffusion paths in a qualitative and quantitative manner (see Fig. 3 for LIPON with isolated tetrahedrons) [7].
Ausblick
Our investigations have shown that the structure of POuN4-u building blocks has a significant influence on the diffusion of vacancies within the LIPON bulk phase. In the next step we will extend the vacancy diffusion to a pseudo-crystalline bulk phase to further increase the dimension of POuN4-u building blocks [8]. Subsequently the interstitial diffusion within those three LIPON structures will be investigated in order to gain further insights into the diffusion mechanisms occurring in LIPON. Our aim is to understand the diffusion processes sufficiently to generate an amorphous-like structure using evolutionary algorithms.