Interface Phenomena in Battery Materials and Exchange Spring Magnets
 Marcel Sadowski
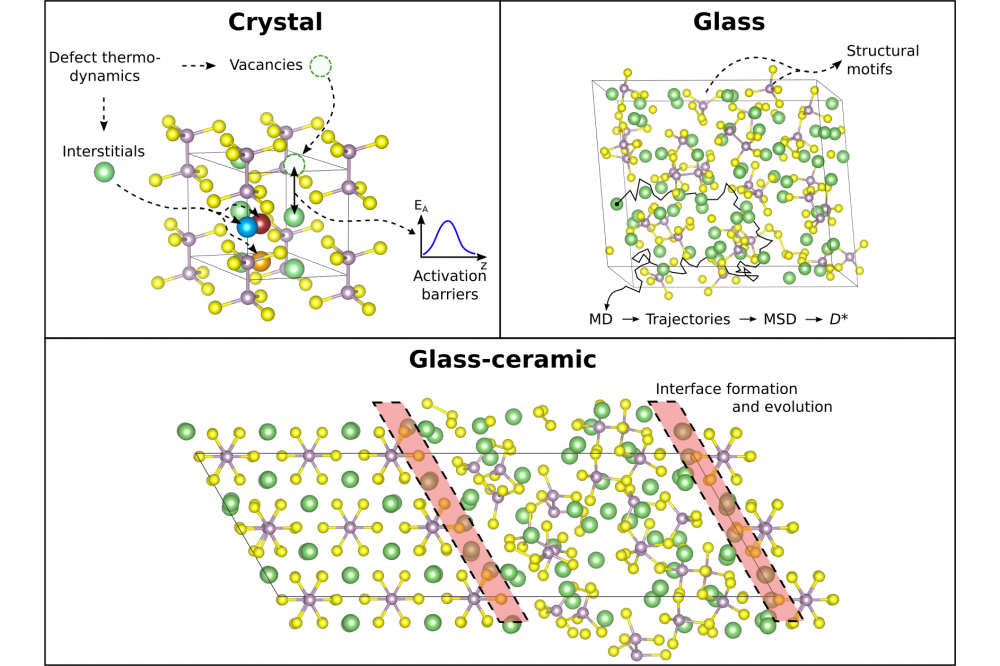
Marcel SadowskiFigure 1: Sketch of our methodology for analyzing lithium thiophosphate glass ceramics. First (top, left) defect thermodynamics of point defects are investigated in crystalline structures. Where applicable, activation barriers can be calculated via nudged elastic band (NEB) calculations. Next, amorphous glass structures are generated with a melt quenching protocol using ab-initio molecular dynamics (AIMD) and the underlying structural motifs can be analyzed (top, right). Structures can be idealized if information from experiments are provided. Furthermore, transport properties like tracer diffusion coefficients (D*) are accessible via AIMD simulations at different temperatures. In the last step (bottom), interfaces can be assembled and their structure, interphases and evolution can be investigated. Defect thermodynamics can be helpful to evaluate the interface stability.
Marcel SadwoskiEinleitung
The project focused on interface phenomena in battery materials and exchange spring magnets but also studied the thermodynamics, defect formation mechanisms and transport properties of the investigated systems. The power of analyzing defect thermodynamics was demonstrated by supporting our experimental partners in understanding the electrochemical processes happening at the interface during sputter deposition of LiPON onto LiCoO2 [1]. Furthermore, we finished our studies on crystalline Li4P2S6 and added a detailed discussion on defect thermodynamics to prove its instability against metallic lithium [2]. We continued our research on solid electrolytes for battery applications with focus on lithium thiophosphates, which can be prepared as ceramics (crystalline), glasses (amorphous) or glass-ceramics (mixed). For latter, the properties of the glassy matrix and the embedded crystallites can vary significantly [3]. Therefore, we started to analyze the glass-ceramic system of 67Li2-33P2S5 (formally Li4P2S7) glasses with embedded Li4P2S6 crystallites. We first generated amorphous structural models of 67Li2-33P2S5 glasses, analyzed their structural motifs and defect thermodynamics, determined diffusion coefficients and activation barriers, repeated the analysis for idealized structural models and compared results to a pseudo-crystalline model [4]. For all structures we observed highly correlated motion of lithium ions and approximately calculated ionic conductivities via tracer diffusion coefficients (D*) instead of using the more appropriate conductivity or jump diffusion coefficient (Dσ) for such cases. The evaluation of Dσ and its statistical (un)certainty will be part of the new project. The average ionic conductivity for 67Li2-33P2S5 glasses was found to be 14 mS/cm at room temperature. Furthermore, our results suggest the instability of glassy Li4P2S7 against metallic lithium but a stable interface between glassy Li4P2S7 and crystalline Li4P2S6.
Methoden
Among quantum-mechanical calculations, Density Functional Theory (DFT) has become an irreplaceable method in nowadays research. DFT studies enable thermodynamic, kinetic and structural investigations of almost any system: bulk, interface or surface models of crystalline or amorphous structures can be analyzed in terms of e.g., thermodynamic stability and electronic/ionic transport or magnetic properties. The only constraint concerns system sizes, which are usually limited to a few hundreds of atoms as standard DFT calculations scale roughly with the cube of the number of involved electrons. Therefore, high-performance computing with largely parallelized calculations are indispensable requirements for executing opperative DFT studies.
Ergebnisse
Transport properties have also been determined for Li4PS4I and we could show that it is indeed a superionic conductor. Experimentally, a lower conductivity was found which we ascribe to grain boundaries, impurities and secondary phases. Therefore, Li4PS4I might be an even better superionic conductor than initially suggested from experiment [5]. Besides battery-related research we analyzed interface phenomena in exchange spring magnets. We showed that the important difference between MnBi- and MnGa-based interfaces are the fact that in the former, a multi-phase (coexistence of crystalline and amorphous phases) and rough interface is formed with higher lattice misfit (6%). In the latter, an epitaxial smooth interface with lower lattice misfit (4.8%) is established [6, 7]. An incoherency in the magnetic hysteresis loop of MnBi interfaces is found, which originates from its interface roughness. Using our DFT and micromagnetic simulations, we found that if a hypothetical smooth interface could have formed in the case of MnBi/FeCo bilayer system, the critical thickness below which the hysteresis loop becomes coherent is between 0.5-1 nm which cannot be detected in the experimental measurements. However, this critical thickness for the case of smooth and epitaxial bilayer system of MnGa/FeCo is increased to 2 nm (to be confirmed with theoretical simulations) due to the increased exchange parameters [6, 7].