Ion Conduction and Defect Formation in Sodium Bismuth Titanate Studied by Density Functional Theory Calculations
 Ion conduction NBT
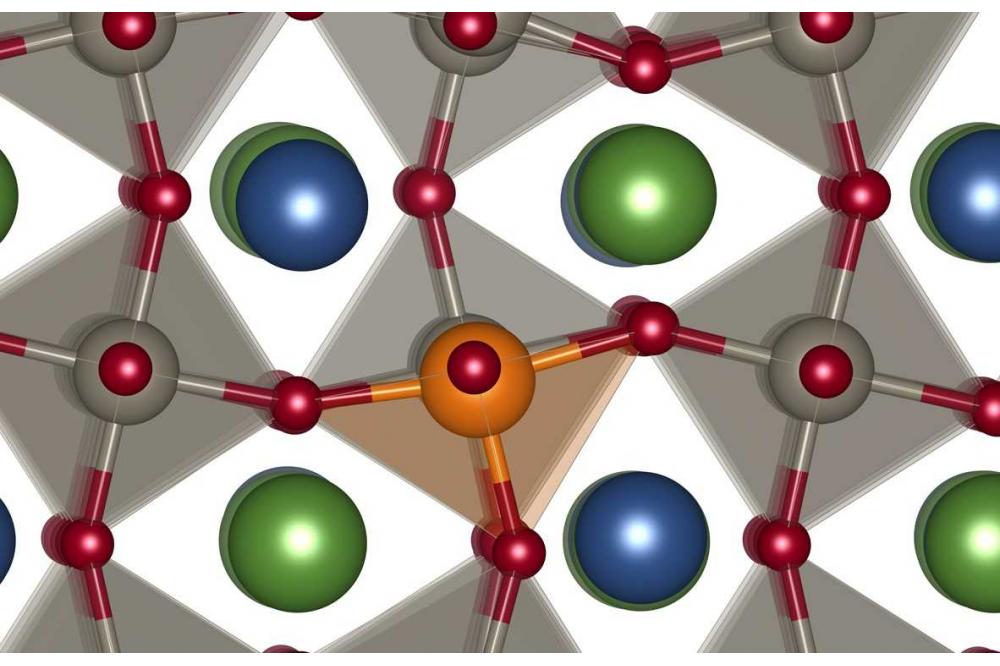
Ion conduction NBTFigure 1: Defect associate of a magnesium acceptor dopant and an oxygen vacancy in NBT with a rock salt like cation order.
Leonie KochEinleitung
Atomistic calculations enable us to observe processes on the micro scale, where common continuum mechanics are not valid anymore. To determine the applicability of NBT (sodium bismuth titanate) as a potential ion conductor for industrial use, we investigate the diffusion of oxygen vacancies within the material and its dependency on external influences. In order to control ionic conductivity, the concentration and mobility of oxygen vacancies can be varied by the incorporation of dopants (substitutional atoms) in the host lattice. However, different elements with various chemical properties can be used for this purpose, leading to a set of design choices. The structural as well as the electronic properties are strongly influenced by the nature of the dopant element as for instance magnesium, iron, or aluminum. However, precisely these characteristics have to be carefully controlled. It has been found that depending on the chemical nature of the dopant, three different types of conductivity mechanisms can be obtained. These are ionic conductivity, mixed ionic and electronic conductivity, and pure electronic conductivity [1]. However, only small amounts of defects can lead to a change of almost three orders of magnitude in the conductivity, which could be detrimental for certain industrial applications. For this reason it is inevitable to understand the influence of mechanical, electrical, and magnetic interaction between the constituents, to optimize the functional properties of NBT based on a physical understanding and apart from a trial and error procedure.
Methoden
Simulations constitute a powerful tool to investigate local scale processes under controlled circumstances. To model the processes at the atomistic level, we apply density functional theory, which describes the spatial distribution of the electrons within the material and is used to calculate the total energy of the system, depending on the electron density. This knowledge theoretically enables us to determine all properties of the material. However, these calculations are computational costly and the amount of time that is needed for the calculations scales with the third power of the number of electrons in the system. Thus a doubling of the size of the cell, increases the computational time by a factor of eight. Therefore, the maximum number of atoms that can be investigated is up to 1000. For these kind of ”large” systems (large for ab-initio calculations) the usage of a high performance computer is essential.
Ergebnisse
From our calculations we could explain why the oxygen migration in NBT behaves differently compared to other known solid electrolytes. In most conductors the conductivity is temperature dependent with an exponentially increase with raising temperature. In NBT the temperature dependence deviates from this known behavior, which has not been understood so far. Recently, we could show that over the interesting temperature range from 100°C to 700°C a phase transition occurs, changing the activation energy for the migration of the oxygen ions. Additionally, binary defect complexes, consisting of an oxygen vacancy and the dopant ion, lead to a trapping of the oxygen vacancies and associated therewith to a temperature dependent effective oxygen vacancy concentration. Furthermore, the dissolution of the complex depends on the structural environment and thus also on the phase transition from a rhombohedral to a tetragonal configuration. However, there are still many unsolved questions, mainly due to the large amount of structural degrees of freedom (symmetry adapted distortion modes). The main challenge we want to conquer is to understand the huge concentration dependency of the conductivity as a function of the dopant.