Computational Investigation of 19F NMR Chemical Shifts in Fe(II) Complexes
 Roman Kornievskii_Computational Investigation of 19F NMR Chemical Shifts in Fe(II) Complexes_Figure1
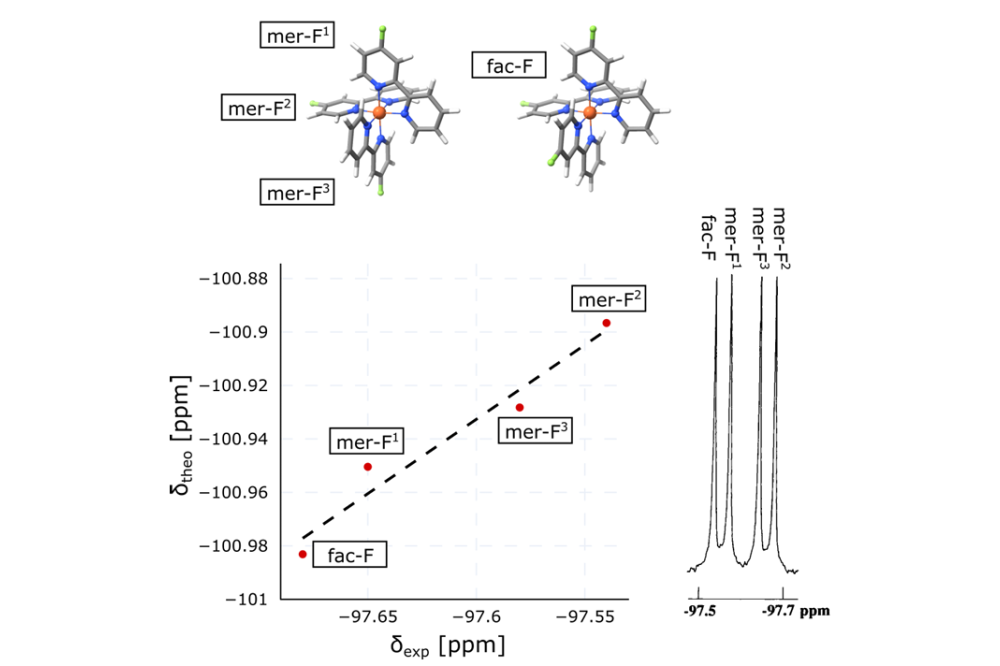
Roman Kornievskii_Computational Investigation of 19F NMR Chemical Shifts in Fe(II) Complexes_Figure1Correlation between theoretical and experimental 19F chemical shifts for the single signal of the facial and the three signals of the meridional isomers of [Fe(bpy-4-F)3]Cl2.
Einleitung
Selective fluorination of organic ligands is a powerful strategy to tune the polarizability and solvation properties of transition metal complexes. For characterising such fluorinated complexes, nuclear magnetic resonance (NMR) spectroscopy offers unique advantages. The 19F nucleus is 100% naturally abundant, has a high gyromagnetic ratio, and covers an exceptionally broad chemical shift range (>400 ppm). Owing to these properties, the readily accessible and highly sensitive 19F NMR chemical shifts have become a valuable probe of the electronic and geometric structures of fluorinated catalytic precursors. The ability to predict 19F NMR parameters computationally is highly desirable for structure elucidation and for linking spectral signatures to the electronic structure. Therefore, the main goal of this project was to establish a set of computational parameters for an accurate prediction of 19F NMR shifts in a set of iron complexes.
Methoden
All quantum-chemical calculations were performed using Kohn–Sham density functional theory (DFT). This method allows one to describe the electronic structures of molecular systems consisting of up to several hundred atoms with sufficient accuracy at relatively low computational cost.
Ergebnisse
A benchmarking study of 19F NMR shifts using global and local hybrid density functionals was performed for a series of fluorinated Fe(II) complexes. It was shown that using a local hybrid density functional instead of global hybrid density functionals significantly improves the agreement with experiment. Furthermore, including scalar relativistic effects or employing a larger basis set was found to introduce only minor changes. Systematic variation of these computational parameters allowed us to establish a protocol for calculating 19F NMR shifts that yields the best agreement with experiment at relatively low computational cost. Accurately calculated chemical shifts also enabled the assignment of very closely spaced 19F NMR signals (within a 0.14 ppm range) to specific atoms in a bipyridine-containing complex that forms facial and meridional geometric configurations (see Figure 1). It is important to note that the assignment of signals within such a small frequency range is not possible from an experimental spectrum alone.
Diskussion
With the available computational power, we were able to perform an extensive benchmarking study consisting of 24 different protocols for 10 iron complexes, with the largest molecular system comprising 163 atoms. Such benchmarking studies are particularly valuable, as they allow future work on similar problems to be conducted more efficiently by establishing a set of computational parameters that yield the best agreement between theory and experiment. For all studied molecular systems, both diamagnetic and paramagnetic, local hybrid density functionals resulted in better agreement with experiment compared to global hybrid functionals. Considering the structural effects, higher mean deviations were observed for flexible molecular structures than for rigid ones. From that, we concluded that for flexible structures multiple conformers can be relevant to obtain reliable 19F NMR shifts computationally, i.e., the theoretical prediction of chemical shifts can be further improved by creating an ensemble of all energetically relevant conformers and calculating an ensemble average of their 19F NMR shifts. To address this, we performed a conformer search for a complex with the largest standard mean deviations among the chemically equivalent fluorine atoms and found ten unique conformers within an energy window of 4 kcal/mol. The chemical shifts of the chemically equivalent fluorine atoms in these conformers exhibit a range of up to 46 ppm. In future work, different sampling methods must be investigated to obtain a method which allows the calculation of relative energies with sufficient accuracy to perform Boltzmann-weighting of the individual conformer shifts to arrive at their ensemble averages. This procedure is expected to significantly improve the agreement of computationally predicted 19F NMR shifts with experiment for flexible molecular systems. Due to the high computational cost of working with an ensemble of conformers instead of a single structure, such calculations can only be performed using a high-performance computer.