Electronic Structure Calculations for Si Anodes in Li Ion Batteries
 Electronic Structure Calculations
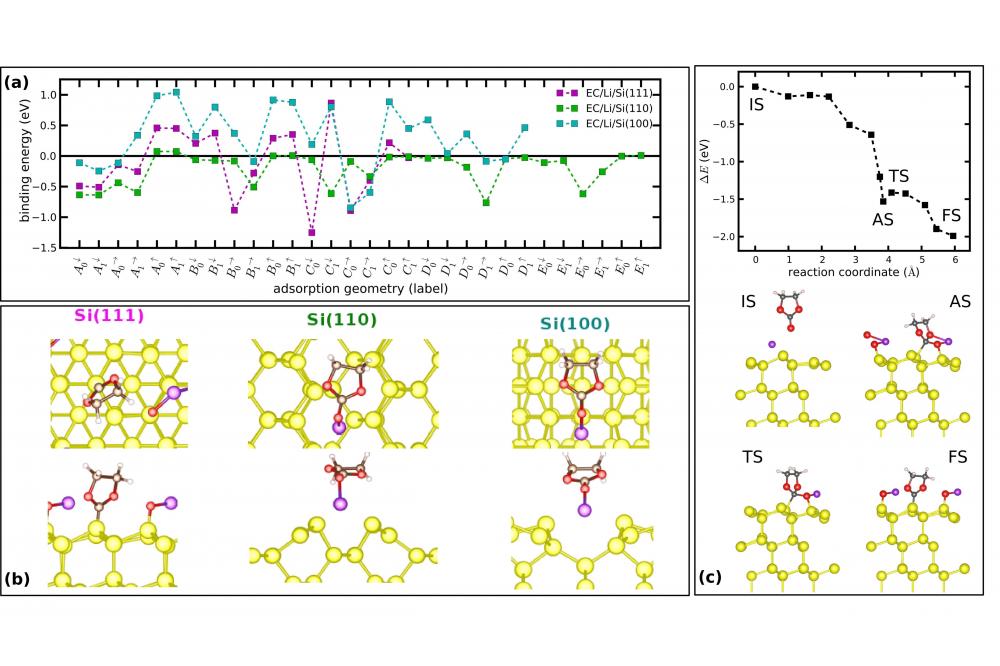
Electronic Structure CalculationsFig. 1. Adsorption and dissociation of EC at different Si surfaces. (a) Energy spectrum of various adsorption geometries (arbitrary labeling); (b) Energetically most favourable adsorption geometries for each surface orientation; (c) Dissociation pathway of EC on Si(111).
Einleitung
Silicon (Si) is one of the most attractive anode materials of Li-ion batteries [1, 2]. However, despite high specific capacity (~4200 mAh/g), large capacity losses and low cyclability have so far prevented commercialization of Si anodes. The main factors leading to capacity losses are the large volume changes during (de)lithiation and the instability of the solid electrolyte interphase (SEI) layer. Formation of the SEI layer typically results from decomposition of electrolyte components during the first (de)lithiation cycles. In subsequent cycles the SEI then acts as a protective layer that prevents the electrolyte from further decomposition and thus stabilizes electrochemical reactions in Li batteries. A detailed understanding of the formation and composition of the SEI on Si is therefore key to functionalize Si anodes for commercial applications.
Methoden
In this work we use density functional theory (DFT) calculations to study the interaction of a particular electrolyte component, namely ethylene carbonate (EC) with the most stable Li-covered Si surfaces [Si(100), Si(110) and Si(111)] [3]. The EC molecule was initially placed on various surface sites with different orientations relative to the surface. Using the projector-augmented planewave code VASP [4] in conjunction with the Perdew-Burke-Ernzerhof [5] parametrization of the exchange-correlation functional, atomic-structure optimizations were performed as to identify favorable adsorption geometries. For details we refer to Ref. 6.
Ergebnisse
Figure 1.a and b show the energy spectrum of all considered adsorption geometries (after relaxation) and the optimized atomic structure of the lowest-energy configurations on Li-covered Si surfaces. On Si(100) and Si(110), the EC molecule stays intact and enters a polar bond (for details on the electronic structure see Ref. 6). On Si(111), the molecule dissociates into a C3H4O2 fragment that is covalently bonded to Si via a C atom and a Li-O dimer. The formation of a Li-O dimer can be seen as a precursor of Li2O, Li2CO3 or Li4SiO4 which have been experimentally identified as constituents of the SEI on Si [7]. Figure 1.c details the dissociation pathway of EC on Si(111). The dissociation barrier is as low as 220 meV. Prior to the dissociation step an energy of 500 meV is released in the adsorption and because of the low thermal conductivity of Si the barrier can easily be overcome. We have also performed ab initio molecular-dynamics simulations to search for dissociation pathways of EC on the other two facets. However, within simulation times of 20 ps no dissociation was observed at room temperature.
Ausblick
This work contributes to an enhanced understanding of the SEI on Si anodes. In particular we find indications that SEI growth starts as soon as a few Li ions start covering the (111) facets while SEI growth on other facets seems to be retarded. In the future this study may be supplemented and extended by exploring other electrolyte components and other relevant anode surfaces.