Structure and Properties of Amorphous LiPON Electrolyte by First-Principles Simulations
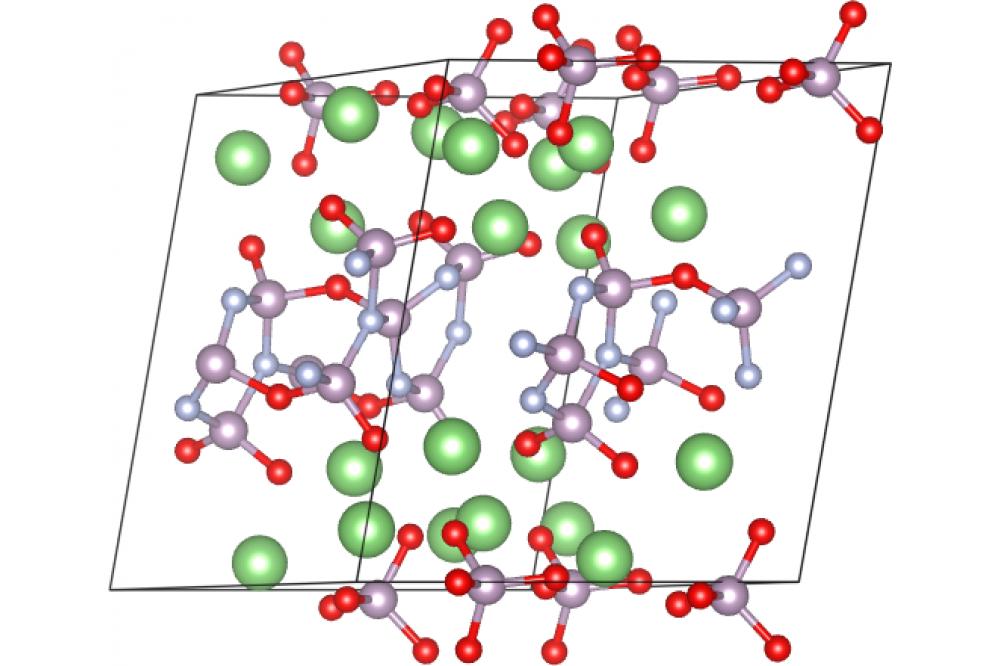
 Structure and Properties of Amorphous 2
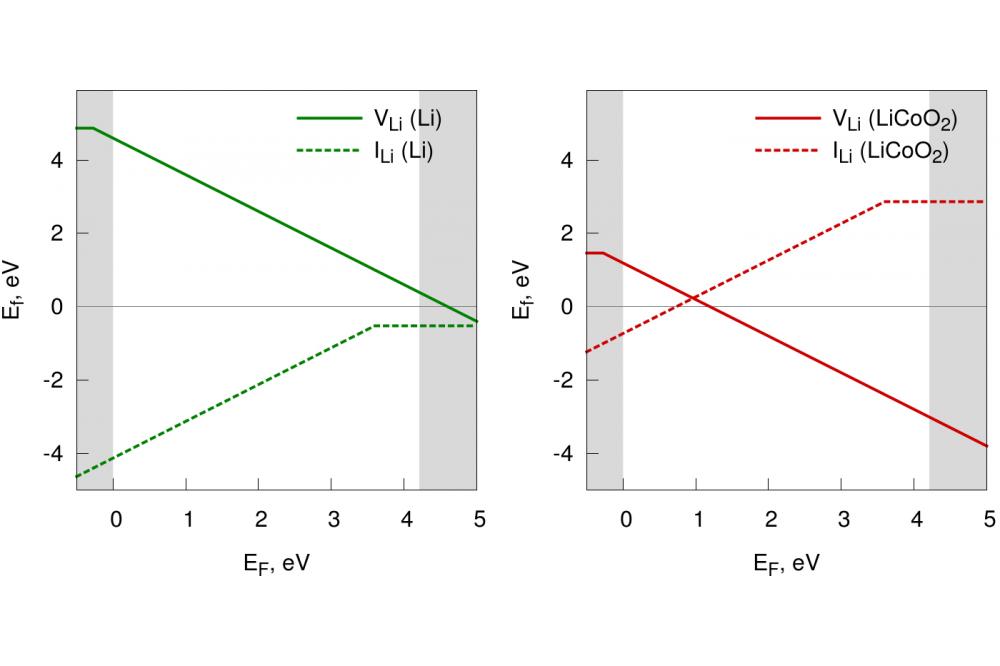
Structure and Properties of Amorphous 2Fig. 2: Defects formation energies as a function of the Fermi level energy with respect to metallic Li (left) and LCO (right). The VBM coincides with EF = 0 and the grey areas represent the valence and conduction bands. The green and red areas locate the stimated positions of Li and LCO Fermi levels, respectively.
Einleitung
Amorphous materials belonging to the family of Lithium Phosphorus Oxynitrides (LiPON) are increasingly popular solid electrolytes for thin-film Li-ion batteries. RF-sputtering of a lithium orthophosphate target in a nitrogen plasma leads to the deposition of amorphous glasses [1]. Besides a fairly high ionic conductivity and very low electronic conductivity, LiPON exhibits remarkable chemical and physical stability to such an extent that its use as a protective layer for cathode materials has been suggested [2]. Rationalization of the transport properties rests on a valid structural model for these glassy structures, whose simulation represents a main challenge from a computational point of view also because of their non-trivial compositions. The motivation for this study is the desire to assess the structural, electronic and transport properties of an amorphous member of the LiPON family with non-trivial composition and cross-linking. The issue of structure prediction for a given composition is a longstanding problem even for the simplest crystalline solids. The problem is further complicated in the case of an amorphous material, as the potential energy landscape that describes the glassy region exhibits a large number of minima of varying depths.
Methoden
We circumvented the problem by using an evolutionary algorithm (USPEX [3]) to find a stable structure for a given composition and subjecting it to ab-initio simulated annealing (VASP [4]) to create disorder (Fig. 1).
Ergebnisse
In this work we present the results of Density Functional Theory calculations. After characterizing the structural and electronic properties of our material, we addressed the issue of ionic conductivity by calculating the defect formation energies of neutral and charged point defects with respect to two different Li reservoirs, namely a typical anode, metallic lithium, and a typical cathode, lithium cobalt oxide (LCO), (Fig. 2). found that charged defects dominate over neutral ones, as expected of an ionic conductor. At the interface with metallic lithium, charged interstitials are favored, whereas at the interface with LCO the relative stability of point defects is reversed and vacancies dominate over interstitials. This trend is consistent with the spontaneous flow of Li ions from the anode (Li) to the cathode (LCO) through the solid electrolyte during the battery discharge. Additionally, the formation of neutral interstitials at the interface with metallic lithium is a competitive process that results in the chemical reduction of LiPON and the disruption of the network compatibly with what experimentally observed [5].
Ausblick
Future studies will address the diffusion of lithium ions across the relevant interfaces with the electrodes and the thermodynamics of side reactions. This tasks imply in turn the investigation of the stability of the interfaces between LiPON and several surfaces of metallic lithium and especially LCO.